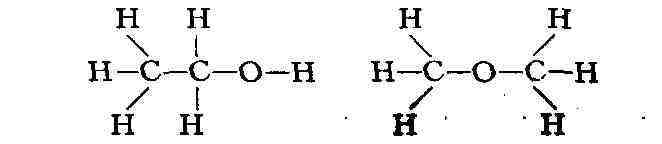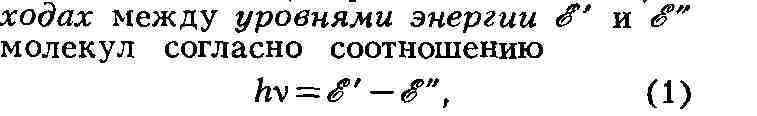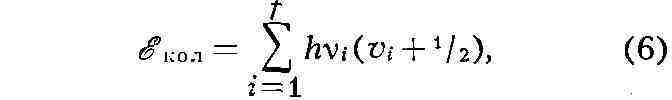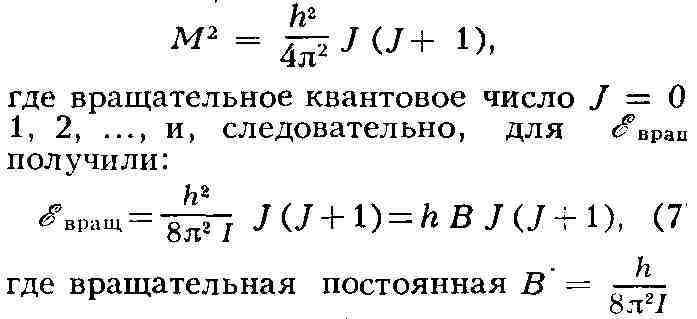![]()
![]()
|
|
|
МОЛЕКУЛА (новолат. molecula, уменьшит, от лат. moles - масса), наименьшая частица вещества, обладающая его хим. свойствами. М. состоит из атомов, точнее - из атомных ядер, окружающих их внутренних электронов и внешних валентных электронов, образующих хим. связи (см. Валентность). Внутр. электроны атомов обычно не участвуют в образовании хим. связей. Состав и строение молекул данного вещества не зависят от способа его получения. В случае одноатомных молекул (напр., инертных газов) понятия М. и атома совпадают. Впервые понятие о М. было введено в химии в связи с необходимостью отличать М. как наименьшее количество вещества, вступающее в хим. реакции, от атома как наименьшего количества данного элемента, входящего в состав М. (Междунар. конгресс в Карлсруэ, I860). Осн. закономерности строения М. были установлены в результате исследования хим. реакций, анализа и синтеза хим. соединений, а также благодаря применению ряда физ. методов. Атомы объединяются в М. в большинстве случаев хим. связями. Как правило, такая связь создаётся одной, двумя или тремя парами электронов, к-рыми владеют сообща два атома. М. может содержать положительно и отрицательно заряженные атомы, т. е. ионы; в этом случае реализуются электростатич. взаимодействия. Помимо указанных, в М. существуют и более слабые взаимодействия между атомами. Между валентно не связанными атомами действуют силы отталкивания. Состав М. выражают формулами химическими. Эмпирич. формула (напр., С2Н6О для этилового спирта) устанавливается на основании атомного соотношения содержащихся в веществе элементов, определяемого хим. анализом, и молекулярной массы. Развитие учения о структуре молекул неразрывно связано с успехами прежде
всего органич. химии. Теория строения органич. соединений, созданная в 60-х гг.
19 в. трудами A.M.. Бутлерова, Ф. А. Кекуле, А. С. Купера и
др., позволила представить строение молекул структурными формулами или
формулами строения, выражающими последовательность валентных хим. связей в М.
При одной и той же эмпирич. формуле могут существовать М. разного строения,
обладающие различными свойствами (явление изомерии). Таковы, напр.,
этиловый спирт С2Н5ОН и диметиловый эфир (СН3)2О.
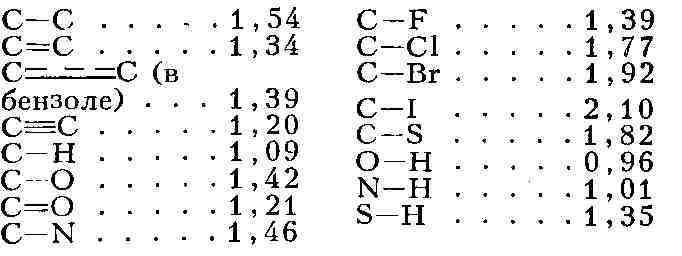
Структурные формулы этих соединений разнятся: В нек-рых случаях изомерные М. быстро превращаются одна в другую и между ними устанавливается динамич. равновесие (см. Таутомерия). В дальнейшем Я. X. Вант-Гофф и независимо франц. химик А. Ж. Ле Бель пришли к пониманию пространственного расположения атомов в молекуле и к объяснению явления стереоизомерии. А. Вернер (1893) распространил общие идеи теории строения на неорганич. комплексные соединения. К нач. 20 в. химия располагала подробной теорией строения М., исходящей из изучения только их хим. свойств. Замечательно, что прямые физ. методы исследования, развитые позднее, в подавляющем большинстве случаев полностью подтвердили структурные формулы химии, установленные путём исследования макроскопич. количеств вещества, а не отдельных М. В физике понятие о М. оказалось необходимым для объяснения свойств газов, жидкостей и твёрдых тел. Прямое экспериментальное доказательство существования М. впервые было получено при изучении броуновского движения (франц. физик Ж. Перрен, 1906). В твёрдом теле М. могут сохранять или не сохранять свою индивидуальность. Так, большинство М. органич. соединений образует молекулярные кристаллы, в узлах решёток к-рых находятся М., связанные одна с другой относительно слабыми силами межмолекулярного взаимодействия. Напротив, в ионных (напр., в случае NaCl) и атомных (алмаз) кристаллах нет отдельных М. и весь кристалл подобен одной М. (см. Кристаллохимия). Структура М. может изменяться при переходе от кристалла к газу. Так, N2Os в газе состоит из единых М., в кристалле - из ионов NO2 и NOa ; газообразный РСl - из М. с конфигурацией тригональной бипирамиды, твёрдый - из октаэдрического иона РС16-и тетраэдрического иона PCl4+. Строение молекул. Геометрическая структура М. определяется равновесным расположением атомных ядер. Энергия взаимодействия атомов зависит от расстояния между ядрами. На очень больших расстояниях эта энергия равна нулю; если при сближении атомов образуется хим. связь, то атомы сильно притягиваются друг к другу (слабое притяжение наблюдается и без образования хим. связи); при дальнейшем сближении атомов действуют электростатич. силы отталкивания атомных ядер; препятствием к сильному сближению атомов является также невозможность совмещения их внутренних электронных оболочек. На рис. 1 показана зависимость потенциальной энергии двухатомной М. от межъядерного расстояния r. Эта энергия минимальна при равновесном расстоянии rа, стремится к нулю при r стремится к бесконечности и возрастает до бесконеч. при r стремящ. к 0. Разность энергий при r0 = r0 и r стремящ. к бесконеч. характеризует энергию связи, энергию диссоциации D (см. табл.). Равновесные расстояния r0 в двухатомных и многоатомных М. и, следовательно, расположение атомных ядер в М. определяются методами спектроскопии, рентгеновского структурного анализа и электронографии, а также нейтронографии, позволяющими получить сведения и о распределении электронов (электронной плотности) в М. Рентгенографич. изучение молекулярных кристаллов даёт возможность установить геометрич. строение очень сложных М., вплоть до М. белков. Косвенную, но весьма детальную информацию о строении сложных М. получают различными спектроскопич. методами, в особенности с помощью спектров ядерного магнитного резонанса (ЯМР). Геометрия простых М., содержащих малое число атомов, также эффективно исследуется методами спектроскопии. Расстояния (в А) Рис. 1. Зависимость потенциальной энергии U двухатомной молекулы (или отдельной химической связи) от межатомного расстояния г (га - равновесное расстояние, D - энергия диссоциации, О, 1, 2,...- уровни энергии колебаний). Равновесные межъядерные расстояния г о и энергии диссоциации D (при 25 °С) некоторых двухатомных молекул
между 2 данными валентно связанными атомами приблизительно постоянны в М.
различных соединений, они уменьшаются с увеличением кратности связи: можно приписать каждому атому в данном валентном состоянии в М.определённый атомный, или ковалентный, радиус в случае ионной связи - ионный радиус, см. Атомные радиусы, Ионные радиусы), характеризующий размеры электронной оболочки атома (иона), образующего хим. связь в М. Представление о приблизительном постоянстве этих радиусов оказывается полезным при оценке межатомных расстояний и, следовательно, при расшифровке структуры М. Длина связи представляет собой сумму соответствующих атомных радиусов. Размер М. как целого, т. е. размер её электронной оболочки, есть величина до
нек-рой степени условная - имеется отличная от нуля, хотя и весьма малая,
вероятность найти электроны М. и на большом расстоянии от её атомных ядер.
практически размеры М. определяются равновесным расстоянием, на к-рое они могут
быть сближены при плотной упаковке М. в молекулярном кристалле и в жидкости. На
больших расстояниях М. притягиваются одна к другой, на меньших - отталкиваются.
Размеры М. поэтому можно найти с помощью рентгеноструктурного анализа
молекулярных кристаллов, порядок величины этих размеров может быть определён из
коэффициентов диффузии, теплопроводности и вязкости газов и из плотности
вещества в конденсированном состоянии. Расстояние, на к-рое могут сблизиться
валентно нe связанные атомы, принадлежащие одной и той же М. или различным М.,
мо-жет быть охарактеризовано средними значениями т. н. ван-дер-ваальсовых
радиусов (в А ): Ван-дер-ваальсовы радиусы существенно превышают ковалентные. Зная величины ван-дер-ваальсовых, ковалентных, атакже ионных радиусов, можно постро-ить наглядные модели М., отражающие форму и размеры их электронных оболочек (рис. 2). Рис. 2. Модели структур некоторых простых молекул (радиусы сфер - ван-дер-ваальсовы). Ковалентные хим. связи в М. расположены под определёнными углами, зависящими
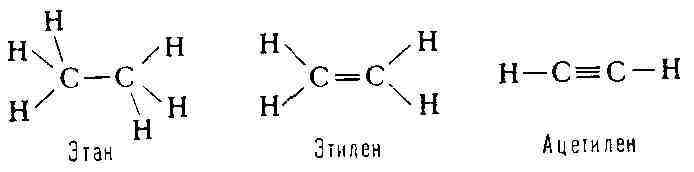
от состояния гибридизации атомных орбиталей (см. Валентность). Так, для
М. насыщенных органич. соединений характерно тетраэдрич. расположение связей,
образуемых атомом углерода; для М. с двойной связью (С = С) - плоское
расположение связей атомов углерода; в М. соединений с тройной связью (С =
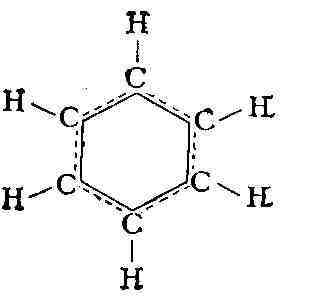
С)-линейное расположение связей: Таким образом, многоатомная М. обладает определённой конфигурацией в пространстве, т. е. определённой геометрией расположения связей, к-рая не может быть изменена без их разрыва. М. характеризуется той или иной симметрией расположения атомов. Если М. не имеет плоскости и центра симметрии, то она может существовать в двух конфигурациях, представляющих зеркальные отражения одна другой (зеркальные антиподы, или стереоизомеры, см. Изомерия). Все важнейшие биологически функциональные вещества в живой природе фигурируют в форме одного определённого стереоизомера. М., содержащие единичные связи, или сигма-связи, могут существовать в различных конформациях, возникающих при поворотах атомных групп вокруг единичных связей. Важные особенности макромолекул синтетич. и биологич. полимеров определяются именно их конформационными свойствами. Взаимодействие атомов в молекуле. Природа хим. связей в М. оставалась загадочной вплоть до создания квантовой механики - классич. физика не могла объяснить насыщаемость и направленность валентных связей. Основы теории хим. связи были созданы В. Гейтлером и нем. учёным Ф. Лондоном в 1927 на примере простейшей молекулы Н2. В дальнейшем теория и методы расчёта были значительно усовершенствованы, в частности на основе широкого применения молекулярных орбиталей метода, и квантовая химия позволяет вычислять межатомные расстояния, энергии М., энергии хим. связей и распределение электронной плотности для сложных М.; при этом расчётные данные хорошо согласуются с экспериментальными. Хим. связи в М. подавляющего числа органич. соединений являются кова-лентными. Напротив, в ряде неорганич. соединений существуют ионные, а также донорно-акцепторные связи (см. Химическая связь), реализуемые в результате обобществления неподелённой пары электронов данного атома. Энергия образования М. из атомов во мн. рядах сходных соединений приближённо аддитивна. Иными словами, в этих случаях можно считать, что энергия М. есть сумма энергий её связей, имеющих постоянные значения в рассматриваемом ряду. Отсюда следует практич. возможность приписать хим. связям приближённо автономные электронные оболочки. Аддитивность энергии М. выполняется не всегда. Яркий пример нарушения аддитивности представляют плоские М. органич. соединений с т. н. сопряжёнными связями, т. е. с кратными связями, чередующимися с единичными. В этих случаях валентные электроны, определяющие кратность связей, т. н. я-электроны, становятся общими для всей системы сопряжённых связей, делокализованными. Такая делокализация электронов приводит к дополнительной стабилизации М. Напр., энергия образования М. 1,3-бутадиена Н2С = СН - СН = СН2 больше ожидаемой по аддитивности на 16,8 кдж/моль (на 4 ккал/моль). Выравнивание электронной плотности вследствие обобществления л-электронов по связям выражается в удлинении двойных связей и укорочении единичных. В правильном шестиугольнике межуглеродных связей бензола (см. формулу) все связи одинаковы и имеют длину, промежуточную между длиной единичной и двойной связи. Сопряжение связей ярко проявляется в молекулярных спектрах (см. ниже). Совр. квантовомеханич. теория хим. связи учитывает частичную делокали-зацию не только я-, но и а-электронов, наблюдающуюся в любых молекулах. Вообще говоря, это не нарушает аддитивности энергий молекул. В подавляющем большинстве случаев суммарный спин валентных электронов в М. равен нулю, т. е. спины электронов попарно насыщены. М., содержащие неспаренные электроны - радикалы свободные (напр., атомный водород Н', метил СН'з), обычно неустойчивы, т. к. при их соединении друг с другом происходит значит, понижение энергии вследствие образования валентных связей. Наиболее эффективным методом изучения строения свободных радикалов является электронный парамагнитный резонанс (ЭПР). Электрические и оптические свойства молекул. Поведение вещества в электрич. поле определяется основными электрич. характеристиками М.- постоянным дипольным моментом и поляризуемостью. Дипольный момент означает несовпадение центров тяжести положит, и от-рицат. зарядов в М., т. е. электрич. асимметрию М. Соответственно М., имеющие центр симметрии, напр. Н2, лишены постоянного дипольного момента; напротив, в НС1 электроны смещены к атому С1 и дипольный момент равен 1,03 D (1,03-10-18 ед. СГС). Поляризуемостью характеризуется способность электронной оболочки любой М. смещаться под действием электрич. поля, в результате чего в М. создаётся индуцированный дипольный момент. Значения дипольного момента и поляризуемости находят экспериментально с помощью измерений диэлектрической проницаемости. В случае аддитивности свойств М. дипольный момент М. может быть представлен суммой дипольных моментов связей (с учётом их направления), то же относится к поляризуемости М. Оптич. свойства вещества характеризуют его поведение в переменном электрич. поле световой волны - тем самым они определяются поляризуемостью М. вещества. С поляризуемостью непосредственно связаны преломление и рассеяние света, оптическая активность и др. явления, изучаемые молекулярной оптикой - разделом физич. оптики, посвящённым изучению оптич. свойств вещества. Магнитные свойства молекул. М. и макромолекулы подавляющего большинства хим.
соединений диамагнитны (см. Диамагнетизм). Магнитная восприимчи- вость М. (х) в ряде органич. соединений может быть выражена как сумма значений х Для отдельных связей; однако аддитивность х выполняется хуже, чем аддитивность поляризуемостей а. И х, и а определяются свойствами внешних электронов М.; эти две величины связаны одна с другой. Парамагнитны М., обладающие постоянным магнитным моментом (см. Парамагнетизм). Таковы М. с нечётным числом электронов во внешней оболочке (напр., NO и любые свободные радикалы), М., содержащие атомы с незамкнутыми (незаполненными) внутр. оболочками (переходные металлы и др.). Магнитная восприимчивость парамагнитных веществ зависит от темп-ры, т. к. тепловое движение препятствует ориентации магнитных моментов в магнитном поле. Строение парамагнитных М. эффективно изучается методом ЭПР. Атомные ядра элементов, у к-рых атомный номер или массовое число нечётны, обладают ядерным спиновым парамагнетизмом. Для таких ядер характерен ядерный магнитный резонанс (ЯМР), спектр к-рого зависит от электронного окружения ядер в М. Поэтому спектры ЯМР служат источником очень подробной информации о строении М., в т. ч. и весьма сложных, напр, белков (см. также Ядерный квадруполъный резонанс, Магнетизм, Магнетохимия). Спектры и строение молекул. Электрич., оптич., магнитные и др. свойства М. в конечном счёте связаны с волновыми функциями и энергиями различных состояний М.; через них выражаются и электрич. дипольный момент, и магнитный момент, и поляризуемость, и магнитная восприимчивость. Прямую информацию о состояниях М. и вероятностях перехода между ними дают молекулярные спектры. Частоты в спектрах, соответствующих вращат. переходам, зависят от моментов инерции М., определение к-рых из спект-роскопич. данных позволяет получить наиболее точные значения межатомных расстояний в М. Общее число линий или полос в колебательном спектре М. зависит от её симметрии. Частоты колебаний, наблюдаемые в спектрах, определяются, с одной стороны, массами атомов и их расположением, с другой - динамикой межатомных взаимодействий. Теория колебаний многоатомных М. соответственно опирается на теорию хим. строения и классическую механику связанных колебаний. Исследование колебательных спектров позволяет сделать ряд выводов о строении М., о межатомных и межмолекулярных взаимодействиях, изучать явления таутомерии, поворотной изомерии. Электронные переходы в М. характеризуют структуру их электронных оболочек, состояние хим. связей. Спектры М., обладающих большим числом сопряжённых связей, характеризуются длинноволновыми полосами поглощения, попадающими в видимую область. Вещества, построенные из таких М., обладают цветностью, к ним относятся все органич. красители. Изучение электронно-колебательных спектров М. необходимо для понимания естественной и магнитной оптич. активности. Молекулы в химии, физике и биологии. Понятие о М.- основное для химии, и большей частью сведений о строении и функциональности М. наука обязана хим. исследованиям. При хим. реакции происходит превращение одних М. в другие. Для такого превращения обычно необходима нек-рая избыточная энергия М.-энергия активации (см. Кинетика химическая). В акте хим. взаимодействия М. проходят через конфигурацию т. н. активированного комплекса, или переходного состояния М. Характер и скорость хим. реакции определяются этим состоянием, в свою очередь зависящим от строения взаимодействующих М. Химия решает две главные задачи, относящиеся к М.,- устанавливает строение М. на основании хим. реакций и, наоборот, на основе строения М. определяет ход реакций. Широкая совокупность важнейших проблем совр. химии, в т. ч. и нерешённых, сводится к теории хим. реакционной способности. Исследование этих проблем требует применения как теоретич. методов квантовой химии, так и экспериментальных данных, получаемых хим. и физ. методами. Физ. явления, определяемые строением и свойствами М., изучаются молекулярной физикой. Термодинамич. свойства любого вещества, построенного из М., в конечном счёте выражаются через значения энергий всех возможных состояний М., находимых из спектроскопич. данных. Строение М. и межмолекулярные взаимодействия ответственны за равновесные свойства вещества. То же относится к неравновесным, кинетич., свойствам. Установление равновесия требует нек-рого времени - времени релаксации. При быстрых изменениях состояния вещества равновесие может не успеть установиться. Эти явления наблюдаются, напр., при прохождении ультразвука через вещество и сказываются на поглощении и дисперсии звуковых волн (см. Молекулярная акустика). Равновесие устанавливается в результате взаимодействия М. при их соударениях в газе и жидкости, в результате поглощения и излучения света и т. д. Время релаксации М. в конденсированной среде существенно зависит от темп-ры, с ростом к-рой увеличивается подвижность М. В ряде случаев М. в жидкости практически утрачивают свою подвижность ещё до кристаллизации: происходит стеклование вещества. Подвижностью М. определяются способность веществ к диффузии, их вязкость, теплопроводность и т. д. Непосредств. изучение подвижности М., определение времён релаксации проводятся методами поглощения и дисперсии электромагнитных волн, ЯМР, ЭПР и др. способами. Равновесные и кинетич. свойства больших цепных М., образующих полимеры (см. Макромолекула), специфичны. Особенности поведения макромолекул определяются прежде всего их гибкостью -способностью находиться в большом числе различных конформаций, возникающих в результате поворотов вокруг единичных связей. Развитие биологии, химии и молекулярной физики привело к построению молекулярной биологии, исследующей осн. явления жизни, исходя из строения и свойств биологически функциональных М. Организм существует на основе тонко сбалансированных химических и нехимических взаимодействий между М. Таким образом, изучение строения и свойств М. имеет фундаментальное значение для естествознания в целом. Лит.: С ы р к и н Я. К., Д я т к и н а М. Е., Химическая связь и строение молекул, М.-Л., 1946; Паулинг Л., Природа химической связи, пер. с англ. М.-Л., 1947; Волькенштейн М. В. Строение и физические свойства молекул М.- Л., 1955; е г о же, Молекулы и жизнь М., 1965; его же, Перекрёстки науки, М. 1972; Кондратьев В. Н., Структура атомов и молекул, 2 изд., М., 1959; К о з м а н У., Введение в квантовую химию, пер. с англ., М., 1960; С л э т е р Дж., Электронная структура молекул, пер. с англ., М., 1965. М. В. Волъкенштейн. МОЛЕКУЛЯРНАЯ АКУСТИКА, раздел физической акустики, в к-ром свойства вещества и кинетика молекулярных процессов исследуются акустич. методами. Осн. методами М. а. являются измерение скорости звука и поглощения звука и зависимостей этих величин от разных физ. параметров: частоты звуковой волны, темп-ры, давления и др. Методами М. а. можно исследовать газы, жидкости, полимеры, твёрдые тела, плазму. Развитие М. а. как самостоят. раздела началось в 30-е годы 20 в., когда было установлено, что во многих веществах при распространении в них звуковых волн имеет место дисперсия скорости звука (см. Дисперсия звука), а поглощение звука не описывается классич. законом, по к-рому коэфф. поглощения пропорционален квадрату частоты. Эти аномалии были объяснены на основании изучения релаксационных процессов (см. Релаксация), что позволило связать нек-рые свойства вещества на молекулярном уровне, а также ряд кинетич. характеристик молекулярных процессов с такими макроскопич. величинами, как скорость и поглощение звука. По скорости звука можно определить такие характеристики вещества, как сжимаемость, отношение теплоёмкостей, упругие свойства твёрдого тела и др., а по поглощению звука - значения сдвиговой и объёмной вязкости, время релаксации и др. В газах, измеряя скорость звука и её зависимость от темп-ры, определяют параметры, характеризующие взаимодействие молекул газа при столкновениях. В жидкости, вычисляя скорость звука на основании той или иной модели жидкости и сравнивая результаты расчёта с опытными данными, в ряде случаев можно оценить правдоподобность используемой модели и определить энергию взаимодействия молекул. На скорость звука влияют особенности молекулярной структуры, силы межмолекулярного взаимодействия и плотность упаковки молекул. Так, напр., увеличение плотности упаковки молекул, появление водородных связей, полимеризация приводят к увеличению скорости звука, а введение в молекулу тяжёлых атомов - к её уменьшению. При наличии релаксац. процессов энергия постулат, движения молекул, к-рую они получают в звуковой волне, перераспределяется на внутр. степени свободы. При этом появляется дисперсия скорости звука, а зависимость произведения коэфф. поглощения на длину волны от частоты имеет максимум на нек-рой частоте, наз. частотой релаксации. Величина дисперсии скорости звука и величина коэфф. поглощения зависят от того, какие именно степени свободы возбуждаются под действием звуковой волны, а частота релаксации, равная обратному значению времени релаксации, связана со скоростью обмена энергией между различными степенями свободы. Т. о., измеряя скорость звука и поглощение в зависимости от частоты и определяя время релаксации, можно судить о характере молекулярных процессов и о том, какой из этих процессов вносит осн. вклад в релаксацию. Этими методами можно исследовать возбуждение колебат. и вращат. степеней свободы молекул в газах и жидкостях, процессы столкновения молекул в смесях различных газов, установление равновесия при химич. реакциях, перестройку молекулярной структуры в жидкостях, процессы сдвиговой релаксации в очень вязких жидкостях и полимерах, различные процессы взаимодействия звука с элементарными возбуждениями в твёрдых телах и др. Анализ акустич. данных для жидкостей обычно проводить труднее, чем для газов, поскольку область релаксации здесь, как правило, лежит в диапазоне более высоких частот, требующем более сложных измерений. В очень вязких жидкостях, полимерах и нек-рых др. веществах в поглощение и дисперсию может давать вклад целый набор релаксац. процессов с широким спектром времён релаксации. Поскольку время релаксации зависит от темп-ры и давления, меняя эти параметры, можно сдвигать по частоте область релаксации. Так, напр., в газе повышение давления газа эквивалентно уменьшению частоты. Это бывает удобно использовать при измерении скорости и поглощения звука, если частота релаксации при нормальных условиях оказывается в том диапазоне частот, к-рый с трудом поддаётся экспериментальному исследованию. Изучение температурных зависимостей скорости и поглощения звука позволяет разделить вклад различных релаксац. процессов. В М. а. для исследований обычно применяется ультразвук: в газах - в диапазоне частот 104-105 гц, а в жидкостях и твёрдых телах - в диапазоне 105-108гц. Это связано как с высоким развитием техники излучения и приёма ультразвука и с большой точностью измерений в этом диапазоне частот, так и с тем, что работа на более низких частотах потребовала бы очень больших объёмов исследуемого вещества, а на более высоких частотах поглощение звука становится столь большим, что многие акустич. методы оказываются неприменимыми. Лит.: Михайлов И. Г., Соловьев В. А., Сырников Ю. П., Основы молекулярной акустики, М., 1964; Физическая акустика, под ред. У. Мэзона, пер. с англ., т. 2, ч. А, М., 1968, т. 4, ч. А и Б, М., 1970; Бергман Л., Ультразвук и его применение в науке и технике, пер. с нем., М., 1956; Herzfeld К. F., Litovitz Т. A., Absorption and dispersion of ultrasonic waves, N. Y.- L., 1959. А.Л. Полякова. МОЛЕКУЛЯРНАЯ БИОЛОГИЯ, наука, ставящая своей задачей познание природы явлений жизнедеятельности путём изучения биол. объектов и систем на уровне, приближающемся к молекулярному, а в ряде случаев и достигающем этого предела. Конечной целью при этом является выяснение того, каким образом и в какой мере характерные проявления жизни, такие, как наследственность, воспроизведение себе подобного, биосинтез белков, возбудимость, рост и развитие, хранение и передача информации, превращения энергии, подвижность и т. д., обусловлены структурой, свойствами и взаимодействием молекул биологически важных веществ, в первую очередь двух главных классов высокомолекулярных биополимеров - белков и нуклеиновых к-т. Отличит, черта М. б. - изучение явлений жизни на неживых объектах или таких, к-рым присущи самые примитивные проявления жизни. Таковыми являются биол. образования от клеточного уровня и ниже: субклеточные орга-неллы, такие, как изолированные клеточные ядра, митохондрии, рибосомы, хромосомы, клеточные мембраны; далее - системы, стоящие на границе живой и неживой природы, - вирусы, в т. ч. и бактериофаги, и кончая молекулами важнейших компонентов живой материи - нуклеиновых кислот и белков. М. б.- новая область естествознания, тесно связанная с давно сложившимися направлениями исследований, которые охватываются биохимией, биофизикой и биоорганической химией. Разграничение здесь возможно лишь на основе учёта применяемых методов и по принципиальному характеру используемых подходов. Фундамент, на к-ром развивалась М. б., закладывался такими науками, как генетика, биохимия, физиология элементарных процессов и т. д. По истокам своего развития М. б. неразрывно связана с молекулярной генетикой, к-рая продолжает составлять важную часть М. б., хотя и сформировалась уже в значит, мере в самостоят- дисциплину. Вычленение М. б. из биохимии продиктовано след, соображениями. Задачи биохимии в основном ограничиваются констатацией участия тех или иных химич. веществ при определённых биологич. функциях и процессах и выяснением характера их превращений; ведущее значение принадлежит сведениям о реакционной способности и об осн. чертах химич. строения, выражаемого обычной химич. формулой. Т. о., по существу, внимание сосредоточено на превращениях, затрагивающих главновалентные химич. связи. Между тем, как было подчёркнуто Л. Полингом, в биологич. системах и проявлениях жизнедеятельности осн. значение должно быть отведено не главновалентным связям, действующим в пределах одной молекулы, а разнообразным типам связей, обусловливающих межмолекулярные взаимодействия (электростатическим, ван-дер-ваальсовым, водородным связям и др.). Конечный результат биохим. исследования может быть представлен в виде той или иной системы химич. уравнений, обычно полностью исчерпываемой их изображением на плоскости, т. е. в двух измерениях. Отличит, чертой М. б. является её трёхмерность. Сущность М. б. усматривается М. Перуцем в том, чтобы истолковать биологические функции в понятиях молекулярной структуры. Можно сказать, что если прежде при изучении биологич. объектов необходимо было ответить на вопрос "что", т. е. какие вещества присутствуют, и на вопрос "где"-в каких тканях и органах, то М. б. ставит своей задачей получить ответы на вопрос "как", познав сущность роли и участия всей структуры молекулы, и на вопросы "почему" и "зачем", выяснив, с одной стороны, связи между свойствами молекулы (опять-таки в первую очередь белков и нуклеиновых к-т) и осуществляемыми ею функциями и, с другой стороны, роль таких отд. функций в общем комплексе проявлений жизнедеятельности. Решающую роль приобретают взаимное расположение атомов и их группировок в общей структуре макромолекулы, их пространственные взаимоотношения. Это касается как отдельных, индивидуальных, компонентов, так и общей конфигурации молекулы в целом. Именно в результате возникновения строго детерминированной объёмной структуры молекулы биополимеров приобретают те свойства, в силу к-рых они оказываются способными служить материальной основой биологич. функций. Такой принцип подхода к изучению живого составляет наиболее характерную, типическую черту М. б. Историческая справка. Огромное значение исследований биологич. проблем на молекулярном уровне предвидел И. П. Павлов, говоривший о последней ступени в науке о жизни - физиологии живой молекулы. Самый термин "М. б." был впервые употреблён англ. учёным У. Астбери в приложении к исследованиям, касавшимся выяснения зависимостей между молекулярной структурой и физич. и биологич. свойствами фибриллярных (волокнистых) белков, таких, как коллаген, фибрин крови или сократительные белки мышц. Широко применять термин "М. б." стали с нач. 50-х гг. 20 в. Возникновение М. б. как сформировавшейся науки принято относить к 1953, когда Дж. Уотсоном и Ф. Криком в Кембридже (Великобритания) была раскрыта трёхмерная структура дезоксирибонуклеиновой кислоты (ДНК). Это позволило говорить о том, каким образом детали данной структуры определяют биологич. функции ДНК в качестве материального носителя наследственной информации. В принципе, об этой роли ДНК стало известно неск. раньше (1944) в результате работ амер. генетика О. Т. Эйвери с сотрудниками (см. Молекулярная генетика), но не было известно, в какой мере данная функция зависит от молекулярного строения ДНК. Это стало возможным лишь после того, как в лабораториях У. Л. Брэгга, Дж. Бернала и др. были разработаны новые принципы рентгеноструктурного анализа, обеспечившие применение этого метода для детального познания пространств, строения макромолекул белков и нуклеиновых кислот. Уровни молекулярной организации. В 1957 Дж. Кендрю установил трёхмерную структуру миоглобина, а в последующие годы это было сделано М. Перуцем в отношении гемоглобина. Были сформулированы представления о различных уровнях пространств, организации макромолекул. Первичная структура - это последовательность отд. звеньев (мономеров) в цепи образующейся молекулы полимера. Для белков мономерами являются аминокислоты, для нуклеиновых кислот -нуклеотиды. Линейная, нитевидная молекула биополимера в результате возникновения водородных связей обладает способностью определённым образом укладываться в пространстве, напр, в случае белков, как показал Л. Полинг, приобретать форму спирали. Это обозначается как вторичная структура. О третичной структуре говорят, когда молекула, обладающая вторичной структурой, складывается далее тем или иным образом, заполняя трёхмерное пространство. Наконец, молекулы, обладающие трёхмерной структурой, могут вступать во взаимодействие, закономерно располагаясь в пространстве относительно друг друга и образуя то, что обозначается как четвертичная структура; её отдельные компоненты обычно наз. субъединицами. Наиболее наглядным примером того, как молекулярная трёхмерная структура определяет биологич. функции молекулы, служит ДНК. Она обладает строением двойной спирали: две нити, идущие во взаимно противоположном направлении (антипараллельно), закручены одна вокруг другой, образуя двойную спираль со взаимно комплементарным расположением оснований, т. е. так, что против определённого основания одной цепи всегда в другой цепи стоит такое основание, к-рое наилучшим образом обеспечивает образование водородных связей: аденин (А) образует пару с тимином (Т), гуанин (Г) - с цитозином (Ц). Такая структура создаёт оптимальные условия для важнейших биологич. функций ДНК: количественного умножения наследственной информации в процессе клеточного деления при сохранении качественной неизменности этого потока генетич. информации. При делении клетки нити двойной спирали ДНК, служащей в качестве матрицы, или шаблона, расплетаются и на каждой из них под действием ферментов синтезируется комплементарная новая нить. В результате этого из одной материнской молекулы ДНК получаются две совершенно тождественные ей дочерние молекулы (см. Клетка, Митоз). Так же и в случае гемоглобина оказалось, что его биологич. функция - способность обратимо присоединять кислород в лёгких и затем отдавать его тканям - теснейшим образом связана с особенностями трёхмерной структуры гемоглобина и её изменениями в процессе осуществления свойственной ему физио-логич. роли. При связывании и диссоциации О2 происходят пространственные изменения конформации молекулы гемоглобина, ведущие к изменению сродства содержащихся в нём атомов железа к кислороду. Изменения размеров молекулы гемоглобина, напоминающие изменения объёма грудной клетки при дыхании, позволили назвать гемоглобин "молекулярными лёгкими". Одна из важнейших черт живых объектов - их способность тонко регулировать все проявления жизнедеятельности. Крупным вкладом М. б. в науч. открытия следует считать раскрытие нового, ранее неизвестного регуляторного механизма, обозначаемого как аллостерический эффект. Он заключается в способности веществ низкой мол. массы - т. н. лигандов - видоизменять специфич. биологич. функции макромолекул, в первую очередь каталитически действующих белков - ферментов, гемоглобина, рецепторных белков, участвующих в построении биологических мембран, в синаптич. передаче (см. Синапсы) и т. д. Три биотических потока. В свете представлений М. б. совокупность явлений жизни можно рассматривать как результат сочетания трёх потоков: потока материи, находящего своё выражение в явлениях обмена веществ, т. е. ассимиляции и диссимиляции; потока энергии, являющейся движущей силой для всех проявлений жизнедеятельности; и потока информации, пронизывающего собой не только всё многообразие процессов развития и существования каждого организма, но и непрерывную череду сменяющих друг друга поколений. Именно представление о потоке информации, внесённое в учение о живом мире развитием М. б., накладывает на неё свой специфический, уникальный отпечаток. Важнейшие достижения молекулярной биологии. Стремительность, размах и глубину влияния М. б. на успехи в познании коренных проблем изучения живой природы справедливо сравнивают, напр., с влиянием квантовой теории на развитие атомной физики. Два внутренне связанных условия определили это революционизирующее воздействие. С одной стороны, решающую роль сыграло обнаружение возможности изучения важнейших проявлений жизнедеятельности в простейших условиях, приближающихся к типу химич. и физич. экспериментов. С другой стороны, как следствие указанного обстоятельства, имело место быстрое включение значит, числа представителей точных наук - физиков, химиков, кристаллографов, а затем и математиков -в разработку биологич. проблем. В своей совокупности эти обстоятельства и обусловили необычайно быстрый темп развития М. б., число и значимость её успехов, достигнутых всего за два десятилетия. Вот далеко не полный перечень этих достижений: раскрытие структуры и механизма биологич. функции ДНК, всех типов РНК и рибосом, раскрытие генетического кода; открытие обратной транскрипции, т. е. синтеза ДНК на матрице РНК; изучение механизмов функционирования дыхательных пигментов; открытие трёхмерной структуры и её функциональной роли в действии ферментов, принципа матричного синтеза и механизмов биосинтеза белков; раскрытие структуры вирусов и механизмов их репликации, первичной и, частично, пространственной структуры антител; изолирование индивидуальных генов; химич., а затем биологич. (ферментативный) синтез гена, в т. ч. человеческого, вне клетки (in vitro); перенос генов из одного организма в другой, в т. ч. в клетки человека; стремительно идущая расшифровка химич. структуры возрастающего числа индивидуальных белков, гл. обр. ферментов, а также нуклеиновых к-т; обнаружение явлений "самосборки" нек-рых биологич. объектов всё возрастающей сложности, начиная от молекул нуклеиновых кислот и переходя к многокомпонентным ферментам, вирусам, рибосомам и т. д.; выяснение аллостерических и др. осн. принципов регулирования биол. функций и процессов. Редукцяонизм и интеграция. М. б. является завершающим этапом того направления в изучении живых объектов, к-рое обозначается как "редукционизм", т. е. стремление свести сложные жизненные функции к явлениям, протекающим на уровне молекул и потому доступным изучению методами физики и химии. Достигнутые М. б. успехи свидетельствуют об эффективности такого подхода. Вместе с тем необходимо учитывать, что в естеств. условиях в клетке, ткани, органе и целом организме мы имеем дело с системами возрастающей степени усложнённости. Такие системы образуются из компонентов более низкого уровня путём их закономерной интеграции в целостности, приобретающие структурную и функциональную организацию и обладающие новыми свойствами. Поэтому по мере детализации познаний о закономерностях, доступных раскрытию на молекулярном и примыкающих уровнях, перед М. б. встают задачи познания механизмов интеграции как линии дальнейшего развития в изучении явлений жизни. Отправной точкой здесь служит исследование сил межмолекулярных взаимодействий - водородных связей, ван-дер-ваальсовых, электростатич. сил и т. д. Своей совокупностью и пространственным расположением они образуют то, что может быть обозначено как "интегративная информация". Её следует рассматривать как одну из гл. частей уже упоминавшегося потока информации. В области М. б. примерами интеграции могут служить явления самосборки сложных образований из смеси их составных частей. Сюда относятся, напр., образование многокомпонентных белков из их субъединиц, образование вирусов из их составных частей - белков и нуклеиновой к-ты, восстановление исходной структуры рибосом после разделения их белковых и нуклеиновых компонентов и т. д. Изучение этих явлений непосредственно связано с познанием осн. феноменов "узнавания" молекул биополимеров. Речь идёт о том, чтобы выяснить, какие сочетания аминокислот - в молекулах белков или нуклеотидов - в нуклеиновых к-тах взаимодействуют между собой при процессах ассоциации индивидуальных молекул с образованием комплексов строго специфичного, наперёд заданного состава и строения. Сюда относятся процессы образования сложных белков из их субъединиц; далее, избирательное взаимовоздействие между молекулами нуклеиновых кислот, напр, транспортными и матричными (в этом случае существенно расширило наши сведения раскрытие генетич. кода); наконец, это образование мн. типов структур (напр., рибосом, вирусов, хромосом), в к-рых участвуют и белки, и нуклеиновые к-ты. Раскрытие соответствующих закономерностей, познание "языка", лежащего в основе указанных взаимодействий, составляет одну из важнейших областей М. б., ещё ожидающую своей разработки. Эту область рассматривают как принадлежащую к числу фундаментальных проблем для всей биосферы. Задачи молекулярной биологии. Наряду с указанными важными задачами М. б. (познанием закономерностей "узнавания", самосборки и интеграции) актуальным направлением науч. поиска ближайшего будущего является разработка методов, позволяющих расшифровывать структуру, а затем и трёхмерную, пространственную организацию высокомолекулярных нуклеиновых к-т. В данное время это достигнуто в отношении общего плана трёхмерной структуры ДНК (двойной спирали), но без точного знания её первичной структуры. Быстрые успехи в разработке аналитич. методов позволяют с уверенностью ждать достижения указанных целей на протяжении ближайших лет. Здесь, разумеется, гл. вклады идут от представителей смежных наук, в первую очередь физики и химии. Все важнейшие методы, использование к-рых обеспечило возникновение и успехи М. б., были предложены и разработаны физиками (ультрацентрифугирование, рентгеноструктурный анализ, электронная микроскопия, ядерный магнитный резонанс и др.). Почти все новые физич. экспериментальные подходы (напр., использование ЭВМ, синхротронного, или тормозного, излучения, лазерной техники и др.) открывают новые возможности для углублённого изучения проблем М. б. В числе важнейших задач практич. характера, ответ на к-рые ожидается от М. о., на первом месте стоит проблема молекулярных основ злокачеств, роста, далее - пути предупреждения, а быть может, и преодоления наследств, заболеваний - "молекулярных болезней". Большое значение будет иметь выяснение молекулярных основ биологич. катализа, т. е. действия ферментов. К числу важнейших совр. направлений М. б. следует отнести стремление расшифровать молекулярные механизмы действия гормонов, токсич. и лекарств, веществ, а также выяснить детали молекулярного строения и функционирования таких клеточных структур, как биологические мембраны, участвующие в регуляции процессов проникновения и транспорта веществ. Более отдалённые цели М. б.-познание природы нервных процессов, механизмов памяти и т. д. Один из важных формирующихся разделов М. б.-т. н. генная инженерия, ставящая своей задачей целенаправленное оперирование генетич. аппаратом (геномом) живых организмов, начиная с микробов и низших (одноклеточных) и кончая человеком (в последнем случае прежде всего в целях радикального лечения наследственных заболеваний и исправления генетич. дефектов). О более обширных вмешательствах в генетич. основу человека речь может идти лишь в более или менее отдалённом будущем, т. к. при этом возникают серьёзные препятствия как технического, так и принципиального характера. В отношении микробов, растений, а возможно, и с.-х. животных такие перспективы весьма обнадёживающи (напр., получение сортов культурных растений, обладающих аппаратом фиксации азота из воздуха и не нуждающихся в удобрениях). Они основаны на уже достигнутых успехах: изолирование и синтез генов, перенос генов из одного организма в другой, применение массовых культур клеток в качестве продуцентов хоз. или мед. важных веществ. Организация исследований по молекулярной биологии. Быстрое развитие М. б. повлекло за собой возникновение большого числа специализированных н.-и. центров. Количество их быстро возрастает. Наиболее крупные: в Великобритании -Лаборатория молекулярной биологии в Кембридже, Королевский ин-т в Лондоне; во Франции - ин-ты молекулярной биологии в Париже, Марселе, Страсбург, Пастеровский ин-т; в США - отделы М. б. в ун-тах и ин-тах в Бостоне (Гарвардский ун-т, Массачусетсский технологич. ин-т), Сан-Франциско (Беркли), Лос-Анджелесе (Калифорнийский тех-нологич. ин-т), Нью-Йорке (Рокфеллеровский ун-т), ин-ты здравоохранения в Бетесде и др.; в ФРГ - ин-ты Макса Планка, ун-ты в Гёттингене и Мюнхене; в Швеции - Каролинский ин-т в Стокгольме; в ГДР - Центр, ин-т молекулярной биологии в Берлине, ин-ты в Йене и Галле; в Венгрии - Биол. центр в Сегеде. В СССР первый специализированный ин-т М. б. был создан в Москве в 1957 в системе АН СССР (см.. Молекулярной биологии институт); затем были образованы: Ин-т биоорганической химии АН СССР в Москве, Ин-т белка в Пущино, Биол. отдел в Ин-те атомной энергии (Москва), отделы М.б.в ин-тах Сиб. отделения АН в Новосибирске, Межфакультетская лаборатория биоорганич. химии МГУ, сектор (затем ин-т) молекулярной биологии и генетики АН УССР в Киеве; значит, работа по М.б. ведётся в Ин-те высокомолекулярных соединений в Ленинграде, в ряде отделов и лабораторий АН СССР и др. ведомств. Наряду с отд. н.-и. центрами возникли организации более широкого масштаба. В Зап. Европе возникла Европ. организация по М. б. (ЕМБО), в к-рой участвует св. 10 стран. В СССР при Ин-те молекулярной биологии в 1966 создан науч. совет по М. б., являющийся координирующим и организующим центром в этой области знаний. Им выпущена обширная серия монографий по важнейшим разделам М. б., регулярно организуются -"зимние школы" по М. б., проводятся конференции и симпозиумы по актуальным проблемам М. б. В дальнейшем науч. советы по М. 6. были созданы при АМН СССР и мн. респ. Академиях наук. С 1966 выходит журнал "Молекулярная биология" (6 выпусков в год). За сравнительно короткий срок в СССР вырос значит, отряд исследователей в области М. б.; это учёные старшего поколения, частично переключившие свои интересы из др. областей; в главной же своей массе это многочисл. молодые исследователи. Из числа ведущих учёных, принявших деятельное участие в становлении и развитии М. б. в СССР, можно назвать таких, как А. А. Баев, А. Н. Белозерский, А. Е. Браунштейн, Ю. А. Овчинников, А. С. Спирин, М. М. Шемякин, В. А. Энгельгардт. Новым достижениям М. б. и молекулярной генетики будет способствовать постановление ЦК КПСС и Сов. Мин. СССР (май 1974) "О мерах по ускорению развития молекулярной биологии и молекулярной генетики и использованию их достижений в народном хозяйстве". Лит.: Вагнер Р., Митчелл Г., Генетика и обмен веществ, пер. с англ.,
М., 1958; Сент-Дьердьи А., Биоэнергетика, пер. с англ., М., 1960; А н ф и н с е
н К., Молекулярные основы эволюции, пер. с англ., М., 1962; Стэнли У., В э л е
н с Э., Вирусы и природа жизни, пер. с англ., М., 1963; Молекулярная генетика,
пер. с англ., ч. 1, М., 1964; В о л ь к е й ш т е й н М. В., Молекулы н жизнь.
Введение в молекулярную биофизику, М., 1965; Гауровиц Ф., Химия н функции
белков, пер. с англ., М., 1965; Б р е с л е р С. Е., Введение в молекулярную
биологию, 3 изд., М. - Л., 1973; Ингрэм В., Биосинтез макромолекул, пер. с
англ., М., 1966; Э н г е л ь г а р д т В. А., Молекулярная биология, в кн.:
Развитие биологии в СССР, М., 1967; Введение в молекулярную биологию, пер. с
англ., М., 1967; У о т с о н Д ж., Молекулярная биология гена, пер. с англ.,
М., 1967; Ф н н е а н Д ж., Биологические ультраструктуры, пер. с англ., М.,
1970; БендоллД ж., Мышцы, молекулы и движение, пер. с англ., М., 1970; И ч а с
М., Биологический код, пер. с англ., М., 1971; Молекулярная биология вирусов,
М., 1971; Молекулярные основы биосинтеза белков, М., 1971; Бернхард С.,
Структура и функция ферментов, пер. с англ., М., 1971; С п и р н н А. С., Гавр
и лова Л. П., Рибосома, 2 изд., М., 1971; Френкель-Конрат X., Химия и
биология вирусов, пер. с англ., М., 1972; Смит К., Хэнеуолт Ф., Молекулярная
фотобиология. Процессы инактивации и восстановления, пер. с англ., М., 1972; X
а р р и с Г., Основы биохимической генетики человека) пер. с англ., М., 1973. МОЛЕКУЛЯРНАЯ ГЕНЕТИКА, раздел генетики и молекулярной биологии, ставящий целью познание материальных основ наследственности и изменчивости живых существ путём исследования протекающих на субклеточном, молекулярном уровне процессов передачи, реализации и изменения генетич. информации, а также способа её хранения. М. г. выделилась в самостоят, направление в 40-х гг. 20 в. в связи с внедрением в биологию новых физич. и химич. методов (рентгеноструктурный анализ, хро-матография, электрофорез, высокоскоростное центрифугирование, электронная микроскопия, использование радиоактивных изотопов и т. д.), что позволило гораздо глубже и точнее, чем раньше, изучать строение и функции отд. компонентов клетки и всю клетку как единую систему. С новыми методами в биологию пришли новые идеи физики и химии, математики и кибернетики. Большую роль в быстром развитии М. г. сыграло перенесение центра тяжести генетич. исследований с высших организмов (эукариотов) - осн. объектов классич. генетики, на низшие (прокариоты) - бактерии и мн. др. микроорганизмы, а также вирусы. Преимущества использования более простых форм жизни для решения генетич. проблем заключаются в быстрой смене поколений у этих форм и возможности изучать одновременно огромное число особей; благодаря этому сильно возрастает разрешающая способность генетич. анализа и повышается его точность. Кроме того, сравнительная простота организации бактерий и особенно вирусов облегчает выяснение молекулярной природы генетич. явлений. Высказываемое иногда мнение о тождестве М. г. и генетики микроорганизмов ошибочно. М. г. изучает молекулярные основы генетич. процессов как у низших, так и у высших организмов и не включает частной генетики прокариотов, занимающей видное место в генетике микроорганизмов. За свою недолгую историю М. г. достигла значит, успехов, углубив и расширив представления о природе наследственности и изменчивости, и превратилась в ведущее и наиболее быстро развивающееся направление генетики. Одно из главных достижений М. г. - выяснение химич. природы гена. Классич. генетика установила, что все наследственные потенции организмов (их генетическая информация) определяются дискретными единицами наследственности - генами, локализованными гл. обр. в хромосомах клеточного ядра, а также в нек-рых органеллах цитоплазмы (пластидах, митохондриях и др.). Однако методы классич. генетики не позволяли вскрыть химич. природу генов, что было отмечено ещё в 1928 выдающимся сов. биологом Н. К. Кольцовым, обосновавшим необходимость изучения механизма наследственности на молекулярном уровне. Первый успех в этом направлении был достигнут при изучении генетич. трансформации у бактерий. В 1944 амер. учёный О. Т. Эйвери с сотрудниками обнаружил, что наследственные признаки одного штамма пневмококков могут быть переданы другому, генетически отличному штамму путём введения в его клетки дезоксирибонуклеиновой кислоты (ДНК), выделенной из первого штамма. Впоследствии подобная генетич. трансформация с помощью ДНК была осуществлена у др. бактерий, а в последнее время - и у нек-рых многоклеточных организмов (цветковые растения, насекомые). Т. о., было показано, что гены состоят из ДНК. Этот вывод был подтверждён опытами с ДНК-содержащими вирусами: для размножения вируса достаточно введения молекул вирусной ДНК в клетку восприимчивого хозяина; все др. компоненты вируса (белки, ли-пиды) лишены инфекционных свойств и генетически инертны. Аналогичные опыты с вирусами, содержащими вместо ДНК рибонуклеиновую кислоту (РНК), показали, что у таких вирусов гены состоят из РНК. Выяснение генетич. роли ДНК и РНК послужило мощным стимулом для изучения нуклеиновых кислот биохимич., физико-химич. и рентгеноструктурными методами. В 1953 амер. учёный Дж. Уотсон и англ. учёный Ф. Крик предложили модель структуры ДНК, предположив, что её гигантские молекулы представляют собой двойную спираль, состоящую из пары нитей, образованных нуклеотидами, расположенными апериодически, но в определённой последовательности. Каждый нуклеотид одной нити спарен с противолежащим нуклеотидом второй нити по правилу комплементарности. Многочисл. экспериментальные данные подтвердили гипотезу Уотсона и Крика. Несколько позже было установлено, что аналогичной структурой обладают молекулы разных РНК, только они большей частью состоят из одной полинуклеотид-ной нити. Дальнейшие работы, в к-рых химич. и физико-химич. методы сочетались с точными генетич. методами (использование разнообразных мутантов, явлений трансдукции, трансформации и т. д.), показали, что разные гены различаются как числом входящих в них пар нуклеотидов (от неск. десятков до полутора тысяч и более), так и строго определённой для каждого гена последовательностью нуклеотидов, в к-рой закодирована генетич. информация. (Принципиально сходную химич. структуру имеют и гены, состоящие из РНК,- у вирусов РНК-типа.) Классич. генетика рассматривала ген как дискретную и неделимую единицу наследственности. Важное значение в пересмотре этой концепции имели работы сов. генетика А. С. Серебровского и его учеников, в 1930-х гг. впервые указавших на возможность делимости гена. Однако разрешающая способность методов классич. генетики была недостаточной для изучения тонкого строения гена. Только с развитием М. г. удалось в 50-60-х гг. решить эту проблему. Мн. работами, проведёнными сначала на бактериях и вирусах, а затем и на многоклеточных организмах, было выяснено, что ген обладает сложным строением: он состоит из десятков или сотен участков - сайтов, способных независимо мутировать и рекомбинировать (см. Мутации, Рекомбинация). Пределом дробим ости гена, а следовательно, и минимальным размером сайта является одна пара нуклеотидов (у вирусов, к-рые содержат одну нить РНК, - один нуклеотид). Установление тонкого строения генов позволило значительно углубить представление о механизме генетич. рекомбинации и закономерностях возникновения генных мутаций, оно способствовало также выяснению механизма функционирования генов. Данные о химич. природе и тонком строении генов позволили разработать методы их выделения. Впервые это было выполнено в 1969 амер. учёным Дж. Бэквитом с сотрудниками для одного из генов кишечной палочки. Затем то же удалось осуществить у нек-рых высших организмов (земноводных). Ещё более значит, успех М. г. - первый химич. синтез гена (кодирующего аланиновую транспортную РНК дрожжей), осуществлённый X. Корана в 1968. Работы в этом направлении ведутся в ряде лабораторий мира. Для внеклеточного синтеза более крупных генов успешно применены новейшие биохимич. методы, основанные на явлении т. н. обратной транскрипции (см. ниже). Используя эти методы, С. Спигелмен, Д. Балтимор, П. Ледер и их сотрудники (США) далеко продвинулись по пути искусств, синтеза генов, определяющих структуру белка в молекулах гемоглобина у кролика и человека. Такие же работы проведены в последнее время и в ряде др. лабораторий, в т. ч. и в СССР. Т. о., М. г. уже выяснила в принципе вопрос о том, как записана и хранится генетич. информация, получаемая потомками от родителей, хотя расшифровка конкретного содержания этой информации для каждого отд. гена требует ещё огромной работы. Установление структуры ДНК открыло возможности для экспериментального исследования биосинтеза молекул ДНК -их репликации. Этот процесс лежит в основе передачи генетич. информации от клетки к клетке и от поколения к поколению, т. е. определяет относит, постоянство генов. Изучение репликации ДНК привело к важному выводу о матричном характере биосинтеза ДНК: для его осуществления необходимо наличие готовой молекулы ДНК, на к-рой, как на шаблоне (матрице), синтезируются новые молекулы ДНК. При этом двойная спираль ДНК раскручивается, и на каждой её нити синтезируется новая, комплементарная ей нить, так что дочерние молекулы ДНК состоят из одной старой и одной новой нити (полуконсервативный тип репликации). Выделен белок, вызывающий раскручивание двойной спирали ДНК, а также ферменты, осуществляющие биосинтез нуклеотидов и их соединение ("сшивание") друг с другом. Несомненно, что в клетке имеются механизмы, регулирующие синтез ДНК. Пути такой регуляции ещё во многом неясны, но очевидно, что она в большой степени определяется генетич. факторами. М. г. достигла выдающегося успеха и в решении важнейшей задачи, сформулированной ещё классич. генетикой, - каким образом ген определяет признак, или как происходит реализация генетич. информации. Предпосылкой послужило сформулированное ещё в 1941 Дж. Бидлом и Э. Тейтемом положение "один ген - один фермент". Это положение позволило поставить вопрос в следующем виде: как гены, т. е., по сути дела, участки молекулы ДНК, определяют химич. структуру и свойства белков, специфич. для данного организма? Раскрытие химич. структуры ДНК и белка дало возможность сопоставить эти два типа биополимеров, что привело к концепции генетического кода, согласно к-рой порядок чередования 4 сортов нуклеотидов в ДНК определяет порядок чередования 20 сортов аминокислот в белковой молекуле. От последовательности расположения аминокислот в белковой молекуле (её первичной структуры)зависят все её свойства. Расшифровка принципов, на к-рых основан генетич. код, была осуществлена в 1962 Ф. Криком с сотрудниками в генетич. опытах с мутантами одного бактериального вируса. Оказалось, что каждая тройка нуклеотидов в цепи ДНК (триплет, кодон) определяет, какая именно из 20 аминокислот займёт данное место в полипептидной цепи синтезируемого белка, т. е. каждый триплет кодирует определённую аминокислоту. Последующие работы позволили полностью рас- шифровать генетич. код и установить нуклеотидный состав всех триплетов, кодирующих аминокислоты, а также состав инициирующего кодона, определяющего начало синтеза данной полипептидной цепи, и трёх терминирующих кодонов, определяющих конец синтеза. Было найдено, что генетич. код универсален для всего живого, т. е. что он один и тот же для любого организма, начиная от вирусов и кончая высшими животными и человеком. Участок молекулы ДНК, составляющий один ген, определяет, как правило, последовательность аминокислот в молекуле одного белка (или в одной полипептидной цепи, если данный белок состоит из неск. таких цепей). Расшифровка генетич. кода сыграла выдающуюся роль в выяснении механизма биосинтеза белка - процесса, включающего перенос заключённой в ДНК генетич. информации на молекулы т. н. информационной, или матричной, РНК (и-РНК). Этот процесс, сущность к-рого составляет синтез и-РНК на матрице ДНК, получил название транскрипции. Информационная РНК связывается затем с особыми клеточными структурами - рибосомами, на к-рых и осуществляется синтез полипептидной цепи в соответствии с информацией, записанной в молекуле и-РНК. Этот процесс синтеза полипептидных цепей при посредстве и-РНК назван трансляцией. Т. о., передача генетич. информации происходит по схеме: ДНК -> РНК -> белок. Это осн. положение (догма), правильность которого установлена мн. исследованиями на различных организмах, получило в 1970 важное дополнение. Американские учёные X. Темин и Д. Балтимор обнаружили, что при репродукции некоторых РНК-содержащих вирусов, вызывающих опухоли у животных, генетическая информация передаётся от РНК вируса к ДНК. Подобная обратная транскрипция осуществляется особыми ферментами, содержащимися в этих вирусах. Явление обратной транскрипции было обнаружено также в нек-рых здоровых клетках животных и человека. Полагают, что обратная транскрипция играет существенную роль в возникновении по крайней мере нек-рых форм злокачественных опухолей и лейкозов, а, возможно, также в процессах дифференцировки при нормальном развитии организмов. Следует подчеркнуть, что открытие обратной транскрипции не противоречит осн. положению М. г. о том, что генетич. информация передаётся от нуклеиновых к-т к белкам, но не может передаваться от белка к нуклеиновым к-там. Замечат. достижение М. г.- раскрытие генетич. механизмов регуляции синтеза белков в бактериальной клетке. Как показали в 1961 франц. учёные Ф. Жакоб и Ж. Моно, биосинтез белка в бактерии находится под двойным генетич. контролем. С одной стороны, молекулярная структура каждого белка детерминируется соответствующим структурным геном, с другой - возможность синтеза этого белка определяется особым геном-регулятором, который кодирует спец. регуляторный белок, способный связываться со специфическим участком ДНК - т. н. оператором - и при этом "включать" или "выключать" функционирование структурных генов, управляемых этим оператором. Система из одного или неск. структурных генов и их оператора составляет т. н. оперон. Способность регуляторных белков связываться с оператором зависит от взаимодействующих с этими белками низкомолекулярных соединений - эффекторов. Эффекторы поступают в клетку извне или синтезируются ею и служат сигналами о необходимости синтеза этой клеткой тех или иных белков или прекращения их синтеза. Регуляторные белки бывают двух типов: белки-репрессоры, к-рые, связываясь с оператором, блокируют синтез белка (негативная регуляция), и белки-активаторы, к-рые, связываясь с оператором, индуцируют синтез белка (позитивная регуляция). При негативной регуляции в одних случаях репрессор до взаимодействия с эффектором находится в активной форме и, связываясь с оператором, препятствует транскрипции структурных генов оперона (а следовательно, и синтезу соответствующих белков). Эффектор переводит репрессор в неактивную форму, оператор освобождается и транскрипция структурных генов (а отсюда и синтез кодируемых ими белков) становится возможной. В др. случаях взаимодействие репрессора с эффектором переводит репрессор в активную форму, в к-рой он способен связаться с оператором, что и приводит к блокированию синтеза белка. При позитивной регуляции, напротив, только активная форма белка-активатора, способная связываться с оператором, обусловливает синтез белка. Активная форма белка-активатора тоже определяется его взаимодействием с эффектором. У многоклеточных организмов генетич. регуляция синтеза белка сложнее и пока изучена недостаточно. Однако ясно, что и здесь большую роль играет обратная связь, подобная описанной у бактерий для системы эффектор - регуляторный белок - оператор, причём сигнальными веществами в ряде случаев служат гормоны. С развитием М. г. более глубоким стало понимание мутационного процесса, т. е. изменения генетической информации. Было показано, что мутации представляют собой либо замены отд. нуклеотидов, либо вставки или выпадения нуклеотидов в молекуле ДНК. Мутации возникают как вследствие случайных ошибок при репликации ДНК, так и в результате повреждающего нуклеиновые к-ты действия различных физич. и химич. агентов -мутагенов; они возникают также из-за изменений т. н. генов-мутаторов, кодирующих ферменты, участвующие в репликации, исправляющие генетич. повреждения и др. Вызываемые мутагенами изменения химич. структуры ДНК либо непосредственно представляют мутации, либо ведут к возникновению мутаций вследствие обусловленных этими изменениями ошибок в ходе последующей репликации ДНК. Значит, доля молекулярных повреждений ДНК, вызываемых мутагенами, не реализуется в мутации, а исправляется (репарируется). Суть явления репарации состоит в том, что у всех организмов имеются гены, кодирующие особые ферменты, способные •"узнавать" повреждённые участки ДНК, "вырезать" их из молекулы и заменять полноценными. Нек-рые из этих ферментов идентифицированы, установлен и механизм их действия, но полного понимания процесса репарации ещё не достигнуто. Изучение репарации открыло новые подходы к исследованию механизма рекомбинации сцепленных (т. е. лежащих в одной хромосоме) генов, представляющей одну из причин комбинативной изменчивости, к-рая наряду с мутациями играет важную роль в эволюции. Классич. генетикой было показано, что рекомбинация сцепленных генов происходит путём обмена гомологичных хромосом участками (кроссинговер), но тонкий механизм такого обмена оставался неизвестным. Экспериментальные данные последних 10-15 лет позволяют рассматривать внутрихромосомную и внутригенную (межсайтовую) рекомбинацию как ферментативный процесс, происходящий при взаимодействии молекул ДНК. Акт рекомбинации осуществляется путём разрывов и соединения в новом сочетании отрезков полинуклеотидных нитей. При этом разрывы с последующим воссоединением могут происходить как одновременно в обеих нитях ДНК (кроссинговер), так и в пределах одной нити (т. н. полукроссинговер). Чтобы имел место кроссинговер, так же как и для репарации, необходимы разрывы, репарационный синтез повреждённых участков и восстановление нарушенных фосфатных связей, осуществляемые соответствующими ферментами. М. г. своими замечательными открытиями оказала плодотворное влияние на все биологич. науки. Она явилась той основой, на к-рой выросла молекулярная биология, значительно ускорила прогресс биохимии, биофизики, цитологии, микробиологии, вирусологии, биологии развития, открыла новые подходы к пониманию происхождения жизни и эволюции органич. мира. Вместе с тем М. г., позволившая глубоко проникнуть в природу важнейших жизненных процессов и успешно продолжающая их исследование, отнюдь не претендует на решение многих, в т. ч. и генетических, проблем, касающихся целостного организма, а тем более совокупностей организмов - популяций, видов, биоценозов и т. д., где преобладают закономерности, изучение к-рых требует иных методов, чем те, какие использует М. г. Достижения М. г., внёсшие огромный теоретич. вклад в общую биологию, несомненно будут широко использованы в практике с. х-ва и медицины (т. н. генная инженерия путём замены вредных генов полезными, в т. ч. искусственно синтезированными; управление мутац. процессом; борьба с вирусными болезнями и злокачественными опухолями путём вмешательства в процессы репликации нуклеиновых к-т и опухолеродных вирусов; управление развитием организмов посредством воздействия на генетич. механизмы синтеза белка и т. д.). Перспективность практич. применения достижений М. г. подтверждается успехами, достигнутыми на модельных объектах. Так, у наиболее изученных в генетич. отношении видов бактерий удаётся получать мутации любого гена, лишать клетку к.-л. гена или привносить в неё желаемый ген извне, регулировать функции мн. генов. Несмотря на то что генетич. свойства клеток эукариотов изучены на молекулярном уровне ещё недостаточно, увенчались успехом первые попытки введения нек-рых генов в клетки млекопитающих с помощью вирусов, осуществлена гибридизация соматических клеток и др. Напр., в 1971 амер. учёный С. Меррилл с сотрудниками, культивируя вне организма клетки человека, больного галактоземией (такие клетки неспособны вырабатывать один из ферментов, необходимых для утилизации молочного сахара, что и является причиной этой тяжёлой наследственной болезни), ввели в эти клетки неинфекционный для них бактериальный вирус, содержащий ген, кодирующий данный фермент. В результате клетки "излечились" - стали синтезировать недостающий фермент и передавать эту способность последующим клеточным поколениям. Уже сейчас данные М. г. используют при создании медикаментов, применяемых для профилактики и лечения новообразований, лейкозов, вирусных инфекций, лучевых поражений, при изыскании новых мутагенов и т. д. Лит.: Вагнер Р., Митчелл Г., Генетика и обмен веществ, пер. с англ., М., 1958; Молекулярная генетика. Сб. ст., пер. с англ., ч. 1, М., 1964; Кольцов Н. К., Наследственные молекулы, "Бюлл. Московского об-ва испытателей природы. Отдел биологический", 1965, т. 70, в. 4, с. 75-104; Б р е с л е р С. Е., Введение в молекулярную биологию, 3 изд., М.-Л., 1973; У о т с о н Д ж.. Молекулярная биология гена, пер. с англ., М., 1967; Гершкович И., Генетика, пер. с англ., М., 1968; X е с и н Р. Б., Энзимология генетических процессов, в кн.: Вопросы молекулярной генетики и генетики микроорганизмов, М., 1968; Р а т н е р В. А., Принципы организации и механизмы моле-кулярно-генетических процессов, Новосибирск, 1972; S t е n t G. S., Molecular genetics, S. F., 1971; E i g e n M., Selforganization of matter and the evolution of biological macromolecules, "Naturwissenschaften", 1971, Jg. 58, H. 10; Baltimore D., Viral RNA-dependent DNA polymerase, "Nature", 1970, v. 226, № 5252; Temin H., Mizutani S., RNA-dependent DNA polymerase in virions of Rous sarcoma virus, "Nature", 1970, v. 226, № 5252; Kacian D. L. [a. o.], In vitro synthesis of DNA components of human genes for globins, "Nature. New Biology", 1972, v. 235, № 58. С. М. Гершензон, Е. И. Черепенко. МОЛЕКУЛЯРНАЯ ДИСТИЛЛЯЦИЯ, способ разделения жидких смесей в высоком вакууме. См. Дистилляция. МОЛЕКУЛЯРНАЯ МАССА, молекулярный вес, значение массы молекулы, выраженное в атомных единицах массы. Практически М. м. равна сумме масс всех атомов, входящих в состав молекулы; умножение М. м. на принятую величину атомной единицы массы (1,66043 ± 0,00031)-10-24 г даёт массу молекулы в граммах. Понятие М. м. прочно вошло в науку после того, как в результате работ С. Канниццаро, развившего взгляды А. Авогадро, были чётко сформулированы различия между атомом и молекулой; уточнению понятия М. м. способствовали открытие Ф. Содди явления изотопии (см. Изотопы) и разработка Ф. Астоном масс-спектрометрического метода определения масс. Понятие М. м. тесно связано с определением молекулы; однако оно приложимо не только к веществам, в к-рых молекулы существуют раздельно (газы, пары, нек-рые жидкости и растворы, молекулярные кристаллы), но и к остальным случаям (ионные кристаллы и др.). За М. м. часто принимают ср. массу молекул данного вещества, найденную с учётом относит, содержания изотопов всех элементов, входящих в его состав. Иногда М. м. определяют не для индивидуального вещества, а для смеси различных веществ известного состава. Так, можно рассчитать, что "эффективная" М. м. воздуха равна 29. М. м.- одна из важнейших констант, характеризующих индивидуальное вещество. М. м. разных веществ сильно различаются между собой. Так, напр., величины М. м. водорода, двуокиси углерода, сахарозы, гормона инсулина соответственно составляют: 2,016; 44,01; 342,296; ок. 6000. М. м. нек-рых биополимеров (белков, нуклеиновых к-т) достигают многих млн. и даже неск. млрд. Величины М. м. широко используются при различных расчётах в химии, физике, технике. Знание М. м. автоматически даёт величину грамм-молекулы (моля), позволяет вычислить плотность газа (пара), рассчитать молярную концентрацию (молярностъ) вещества в растворе, найти истинную формулу соединения по данным о его составе и т. д. Экспериментальные методы определения М. м. разработаны гл. обр. для газов (паров) и растворов. В основе определения М. м. газов (паров) лежит Авогадро закон. Известно, что объём 1 моля газа (пара) при нормальных условиях (0 °С, 1 атм) составляет ок. 22,4 л; поэтому, определив плотность газа (пара), можно найти число его молей, а следовательно, найти и М. м. В случае растворов для определения М. м. чаще всего используют криоскопический и эбулиоскопический методы (см. Криоскопия и Эбулиоскопия). Экспериментальные методы дают сведения о ср. значении М. м. вещества. Оценку М. м. отд. молекул можно проводить методом масс-спектрометрии. М. м. являются важной характеристикой высокомолекулярных соединений -полимеров, определяющей их физ. (и технологические) свойства. Макромолекулы. полимеров образуются повторением сравнительно простых звеньев (групп атомов); число мономерных звеньев, входящих в состав различных молекул одного и того же полимерного вещества, различно, вследствие чего М. м. макромолекул таких полимеров также неодинакова. Поэтому при характеристике полимеров обычно говорят о ср. значении М. м.; эта величина даёт представление о ср. числе звеньев в молекулах полимера (о степени полимеризации ). Полное описание размеров молекул полимера даёт функция распределения по М. м. (молекулярно-массовое распределение); эта функция позволяет найти долю молекул (определённого размера) данного полимерного вещества, М. м. к-рых лежат в заданном интервале масс (от М до М + ДМ). На практике обычно определяют ср. М. м. полимера, исследуя тем или иным методом его раствор. Свойства растворов могут зависеть от числа молекул, находящихся в растворе (при этом разные по массе молекулы ведут себя совершенно одинаково), от массовой (весовой) концентрации раствора (в этом случае одна большая молекула производит такой же регистрируемый эффект, как и неск. малых) и от др. факторов. Если полимер состоит из неодинаковых молекул, то ср. значения М. м., измеренные разными способами, будут различны. Так, понижение темп-ры замерзания (повышение темп-ры кипения) разбавленного раствора зависит только от числа содержащихся в нём молекул, а не от их размеров, поэтому криоскопич. и эбулиоскопич. методы позволяют находить среднечисленную М. м. полимера ("простое" среднее). Интенсивность света, рассеянного раствором полимера, зависит от массы вещества, находящегося в растворе, а не от числа молекул; поэтому метод, основанный на измерении интенсивности рассеянного света, используется для определения величины М. м. полимера, усреднённой по массе. Др. методы (седиментационного равновесия, вискозиметрический и т. д.) позволяют найти иные ср. значения М. м. полимеров. Сравнивая ср. величины М. м., определённые разными методами, можно сделать вывод о молекулярно-массовом распределении. В простейшем случае, когда среднечисленная М. м. полимера совпадает со значением М. м., усреднённой по массе, можно сделать вывод, что полимер состоит из одинаковых молекул (т. е. монодисперсен). Лит.: Некрасов Б. В., Основы общей химии, т. 1, М., 1973; Гуггенгейм Э.А. и Пру Дж., Физико-химические расчёты, пер. с англ., М., 1958; Губен-Вейль, Методы органической химии, т. 2, М., 1967. См. также лит. при ст. Макромолекула. С. С. Бердоносов. МОЛЕКУЛЯРНАЯ ОПТИКА, раздел оптики, в к-ром изучаются процессы взаимодействия оптического излучения с веществом, существенно зависящие от атомно-молекулярной структуры вещества. М. о. устанавливает связь между характером единичных актов взаимодействия световой волны с частицами (молекулами, атомами, ионами) и макроскопич. параметрами состоящей из этих частиц среды (напр., её показателем преломления). С этой точки зрения в М. о. рассматриваются дисперсия света, преломление света и - наиболее широко - рассеяние света. Изучение распространения света в кристаллах, обладающих естественной оптической анизотропией, составляет предмет кристаллооптики. Оптическая анизотропия в изотропных от природы средах может вызываться действием на них различных внешних полей: электрического (см. Керра эффект, Поккельса эффект), магнитного (см. Коттона - Мутона эффект), поля механич. или гидродинамич. сил (явления фотоупругости и двойного лучепреломления в потоке жидкости). В средах, для к-рых характерна оптическая активность (как естественная, так и возникающая при наложении внешнего магнитного поля, см. Фарадея эффект), происходит вращение плоскости поляризации света. Все эти явления, рассматриваемые в М. о., дают ценную информацию о свойствах веществ и строении составляющих их частиц. Процесс взаимодействия световой волны с частицами вещества определяется гл. обр. поляризуемостью этих частиц (см. Поляризуемость атомов, ионов и молекул). Объяснение большинства молекулярно-оптических (МО) явлений дала уже классич. электронная теория, однако для их полного теоретич. истолкования необходима квантовая механика, к-рая позволяет связать МО постоянные со значениями уровней энергии молекул и вероятностями квантовых переходов между этими уровнями (см. Молекула, Молекулярные спектры). Приложения М. о. разнообразны и расширились с появлением источников мощного когерентного излучения - лазеров, Наиболее широко методы М. о. применяются для исследования структуры и характеристик отд. молекул. Изучение света, рассеиваемого различными средами даёт сведения (часто уникальные) о строении этих сред - жидкостей, кристаллов высокомолекулярных соединений, атмосферных образований (облаков, туманов и пр.), а также об особенностях теплового движения частиц в средах. М. о. тесно связана с молекулярной спектроскопией Разрабатываются перспективные М. о методы исследования космич. тел и сред. Лит.: Волькенштейн М. В., Молекулярная оптика, М.- Л., 1951; Б о р н М., Вольф Э., Основы оптики, пер. с англ. 2 изд., М., 1973; Волькенштейн М. В. Строение н физические свойства молекул М. -Л., 1955. В. А. Замков. МОЛЕКУЛЯРНАЯ РЕФРАКЦИЯ, см Рефракция молекулярная. МОЛЕКУЛЯРНАЯ ФИЗИКА, раздел физики, в к-ром изучаются физич. свойства тел в различных агрегатных состояниях на основе рассмотрения их микроскопич. (молекулярного) строения. Задачи М. ф. решаются методами физич. статистики, термодинамики и физич. кинетики, они связаны с изучением движения и взаимодействия частиц (атомов, молекул, ионов), составляющих физич. тела. Атомистич. представления о строении вещества, высказанные ещё философами древности (см. Атомизм), в нач. 19 в. были с успехом применены в химии (Дж. Дальтон, 1801), что в значит. мере содействовало развитию М. ф. Первьм сформировавшимся разделом М. ф. была кинетическая теория газов. В результате работ Дж. Максвелла (1858-60), Л. Больцмана (1868) и Дж. Гиббсс, (1871 -1902), развивавших молекулярно-кинетич. теорию газов, была создана классич. статистическая физика. Количественные представления о взаимодействии молекул (молекулярных силах) начали развиваться в теории капиллярных явлений. Классич. работы в этой области А. Клеро (1743), П. Лапласа (1806), Т. Юнга (1805), С. Пуассона, К. Гаусса (1830-31), Дж. Гиббса (1874-1878), И. С. Громеки (1879, 1886) и др. положили начало теории поверхностных явлений. Межмолекулярные взаимодействия были учтены Я. ван дер Ваальсом (1873) при объяснении физич. свойств реальных газов и жидкостей. В нач. 20 в. М. ф. вступает в новый период своего развития, характеризующийся доказательствами реального строения тел из молекул в работах Ж. Перрена и Т. Сведберга (1906), М. Смолуховского и А. Эйнштейна (1904-06), касающихся броуновского движения микрочастиц, и исследованиями молекулярной структуры веществ. Применение для этих целей дифракции рентгеновских лучей в работах М. Лауэ (1912), У. Г. Брэгга и У. Л. Брэгга (1913), Г. В. Вульфа (1913), А. Ф. Иоффе (1924), В. Стюарда (1927-31), Дж. Бернала (1933), В. И. Данилова (1936) и др., а в дальнейшем и дифракции электронов и нейтронов дало возможность получить точные данные о строении кристаллич. твёрдых тел и жидкостей. Учение о межмолекулярных взаимодействиях на основании представлений квантовой механики получило развитие в работах М. Борна (1937-39), П. Дебая (30-е гг. 20 в.), Ф. Лондона (1927) и В. Гейтлера (1927). Теория переходов из одного агрегатного состояния в другое, намеченная в 19 в. Я. ван дер Ваальсом и У. Томсоном (Кельвином) и развитая в работах Дж. Гиббса, Л.Ландау (1937), М. Фольмера (30-е гг. 20 в.) и их последователей, превратилась в совр. теорию образования новой фазы - важный самостоятельный раздел М. ф. Объединение статистич. методов с совр. представлениями о структуре веществ в работах Я. И. Френкеля (1926 и др.), Г. Эйринга (1935-36), Дж. Бернала и др. привело к М. ф. жидких и твёрдых тел. Круг вопросов, охватываемых М. ф., очень широк. В ней рассматриваются строение газов, жидкостей и твёрдых тел, их изменение под влиянием внешних условий (давления, темп-ры, электрич. и магнитного полей), явления переноса (диффузия, теплопроводность, внутр. трение), фазовое равновесие и процессы фазовых переходов (кристаллизация и плавление, испарение и конденсация и др.), критическое состояние вещества, поверхностные явления на границах раздела различных фаз. Интенсивное развитие М. ф. привело к выделению из неё ряда крупных самостоятельных разделов, таких, напр., как статистич. физика, кинетика физическая, физика твёрдого тела, физическая химия, молекулярная биология. Совр. наука и техника используют всё большее число новых веществ и материалов. Выявившиеся особенности строения этих тел привели к развитию различных науч. подходов к их исследованию. Так, на основе общих теоретич. представлений М. ф. получили развитие такие спец. области науки, как физика металлов, физика полимеров, физика плазмы, кристаллофизика, физико-химия дисперсных систем и поверхностных явлений, теория тепло- и массопереноса. Сюда же можно отнести также новую область науки -Физико-химическую механику, к-рая составляет теоретич. основу совр. материаловедения, указывая пути создания технически важных материалов с требуемыми физич. свойствами. При всём различии объектов и методов исследования здесь сохраняется, однако, осн. идея М. ф.: описание макроскопич. свойств вещества, исходя из особенностей микроскопич. (молекулярной) картины его строения. Лит.: Кикоин И. К. и Кикоин А. К., Молекулярная физика, М., 1963; Гиршфельдер Дж., Кертисс Ч. и Б е р д Р., Молекулярная теория газов и жидкостей, пер. с англ., М., 1961; Френкель Я. И., Собр. избр. трудов, т. 3. - Кинетическая теория жидкостей, М.- Л., 1959; Франк-Каменецкий Д. А. Диффузия и теплопередача в химической кинетике, 2 изд., М., 1967; К и т т е л ь Ч., Введение в физику твёрдого тела, пер. с англ., М., 1957; Лихтман В.И., Щукин Е.Д., Р е б и н д е р П. А., Физико-химическая механика металлов, М., 1962. П. А. Ребиндер, Б. В. Дерягин, Н. В. Чураев. МОЛЕКУЛЯРНАЯ ЭЛЕКТРОНИКА, первоначальное название одного из направлений микроэлектроники. Вместо термина "М. э.", получившего нек-рое распространение в 60-е гг. 20 в., с нач. 70-х гг. применяют другой термин -функциональная электроника. МОЛЕКУЛЯРНОЕ ТЕЧЕНИЕ, течение разреженного газа (молекул, атомов, ионов или электронов), при к-ром свойства потока существенно зависят от беспорядочного движения молекул, в отличие от течений, где газ рассматривается как сплошная среда. М. т. имеет место при полёте тел в верхних слоях атмосферы, в вакуумных системах и т. д. При М. т. молекулы (или др. частицы) газа участвуют, с одной стороны, в поступательном движении всего газа в целом, а с другой- двигаются хаотически и независимо друг от друга. Причём в любом рассматриваемом"" объёме молекулы газа могут иметь самые различные скорости. Поэтому основой теоретич. рассмотрения М. т. является кинетическая теория газов. Макроскопич. свойства невязкого, сжимаемого, изоэнтропического течения удовлетворительно описываются простейшей моделью молекул в виде упругих гладких шаров, к-рые подчиняются максвелловскому закону распределения скоростей (см. Максвелла распределение). Для описания вязкого, неизоэнтропич. М. т. необходимо пользоваться более сложной моделью молекул и функцией распределения, к-рая несколько отличается от функции распределения Максвелла. М. т. исследуются в аэродинамике разреженных газов. Лит.: Паттерсон Г. Н., Молекулярное течение газов, пер. с англ., М., 1960; Чепмен С., Каулинг Т., Математическая теория неоднородных газов, пер. с англ., М., 1960; Аэродинамика разреженных газов. Сб., под ред. С. В. Валландера, Л., 1963; Коган М. Н., Динамика разреженного газа, М., 1967. Л.В. Козлов. МОЛЕКУЛЯРНОЙ БИОЛОГИИ ИНСТИТУТ АН СССР, головное н.-и. учреждение в области молекулярной биологии. Организован в 1957 (до 1965 -Ин-т радиац. и физико-химич. биологии). Основатель и директор ин-та - В. А. Энгельгардт. Осн. направления н.-и. работ: передача и реализация наследственной информации, молекулярные механизмы биосинтеза белка, химич. и физич. основы действия ферментов, связь структуры нуклеиновых к-т и белков с их функциями в клетке, макромолекулярная организация хромосом, разработка физич. методов исследования макромолекул. В М. б. и. расшифрована первичная структура двух транспортных рибонуклеиновых к-т (т-РНК); экспериментально обоснована и сформулирована теория регуляции функционирования генома у высших организмов; впервые в СССР определена последовательность аминокислот в крупной молекуле белка-фермента - аспартатамино-трансферазы (совм. с Ин-том биоорганической химии им. М. М. Шемякина АН СССР); предложены новые подходы к изучению строения активных центров ферментов (ингибиторный анализ) и функциональных участков РНК (метод "разрезанных молекул"); разработаны новые методы структурных исследований белков и нуклеиновых к-т. К нач. 1973 в М. б. и. было 13 лабораторий. Совм. с Советом по проблемам молекулярной биологии АН СССР ин-т организует междунар. совещания и симпозиумы. Труды сотрудников М. б. и. публикуются в журналах: "Молекулярная биология" (с 1967), "Биохимия" (с 1936), "Цитология" (с 1959), "Доклады АН СССР" (с 1933), "Биофизика" (с 1956), "Biochimica et Biophysica Acta" ((N. Y.-Amst., с 1947), "FEBS Letters" (Amst., с 1968), "European Journal of Biochemistry" (В., с 1967), в сборниках и в виде монографий. Лит.: Институт молекулярной биологии, М., 1971. М. Я. Тимофеева. "МОЛЕКУЛЯРНЫЕ БОЛЕЗНИ", врождённые ошибки метаболизма, заболевания, обусловленные наследственными нарушениями обмена веществ. Термин "М. б." предложен амер. химиком Л. Полингом. В нач. 20 в. англ, врач А. Э. Гаррод, изучая ряд наследственных заболеваний, предположил, что они возникают в результате пониженной активности или полного отсутствия фермента, контролирующего определённый этап обмена веществ. Так, появление гомогентизиновой к-ты в моче больных алькаптонурией обусловлено отсутствием окисляющего её фермента (впоследствии выяснилось, что в этом случае образуется неактивная форма фермента); альбинизм вызван блокадой образования пигментов меланинов вследствие недостаточности одного из необходимых ферментов - тирозиназы и т. д. Идеи Гаррода получили всеобщее признание и конкретную химич. интерпретацию спустя неск. десятилетий. Решающими для понимания механизмов возникновения "М. б." оказались исследования изменений биосинтеза у микроорганизмов, возникающих при замене нормального гена мутантным. Каждый нормальный ген определяет (кодирует) синтез, как правило, строго определённого фермента, т. е. нормального белка (см. Белки, Генетический код). Изучение биохимич. мутантов (работы гл. обр. амер. генетиков Дж. Бидла и Э. Тейтема, 1941) показало, что мутация гена приводит к отсутствию фермента или изменению его активности, т. е. белок либо не синтезируется вообще, либо синтезируется с изменённой первичной структурой (иной последовательностью аминокислот в полипептидной цепи). Изменение первичной структуры белка (ферментного, структурного, плазмы крови), по-видимому, не влияет на его свойства ("молчащие" мутации). Однако в ряде случаев (напр., при изменении активного центра фермента) происходит изменение свойств, а следовательно, и функций белка. Т. о., все "М. б." связаны либо с утратой к.-л. нормального белка, либо с изменением его ферментативных или физ.-хим. свойств. Поскольку каждый фермент контролирует определённую реакцию обмена веществ, его отсутствие или неспособность осуществлять свою функцию приводят к остановке нормального пути метаболизма на стадии биосинтеза вещества, являющегося субстратом этого фермента. Заболевание развивается в результате недостатка в организме конечного продукта, синтез к-рого блокирован, либо в результате накопления предшественника блокированной реакции, избыток которого нарушает обменные процессы. "М. б." включают расстройства обмена аминокислот (аминоацидурии), углеводов (гликозурии), липидов (липидозы и лейкодистрофии), пуринов, пиримидинов. Всего известно св. 1000 "М. б.". Частота каждой из "М. б." относительно невелика: одна из самых распространённых "М. б."-фенилкетонурия - встречается со ср. частотой 1 : 10 000. Некоторые из наследственных нарушений обмена не влекут за собой клинич. последствий (напр., неспособность ощущать вкус или запах определённых веществ), другие же протекают очень тяжело. Ряд "М. б." проявляется лишь при воздействии провоцирующих факторов внешней среды. При своевременном диагнозе нек-рые "М. б." поддаются эффективному предупреждению и лечению. Поскольку эффект мутантного гена осуществляется преим. в форме изменения строго определённого биосинтеза, установление наследственного характера болезни (с помощью различных методов биохимич. анализа) открывает возможность воздействия на всю цепь реакций, ведущих к биохимич. и физиологич. аномалиям. Заместительная терапия применяется при гормональных заболеваниях (сахарный диабет лечат инсулином, наследственные формы гипотиреоза - гормоном щитовидной железы). Для лечения ряда "М. б." эффективно применение ограничительных диет, из к-рых изъято вещество (аминокислота, углевод), накапливающееся в организме. Предупреждение "М. б." осуществляется путём медико-генетич. консультирования семей, в к-рых выявлены носители "М. б.". Для ряда "М. б" разработаны методы ранней (в т. ч. внутриутробной) диагностики. Нек-рые "М. б.", напр. т. н. эритроцитопатии, широко распространились в Африке и странах Средиземноморья, т. к. превращают аномальный эритроцит в среду, неблагоприятную для развития малярийного плазмодия (см. Гемоглобинопатии, Малярия). См. также Генетика медицинская, Генетика человека, Медико-генетическая консультация. Лит.: Цукеркандль Э., П о л и н г Л., Молекулярные болезни, эволюция и генная разнородность, в сб.: Горизонты биохимии, пер. с англ., М., 1964; Э ф р о и м с о н В. П., Введение в медицинскую генетику, 2 изд., М., 1968; "Журнал Всесоюзного химического общества им. Д. И. Менделеева", 1970, т. 15, № 6 (посвящен биохимии наследственных болезней человека); Проблемы медицинской генетики, М., 1970; Gar-rod А. Е., Inborn errors of metabolism, L., 1963; The metabolic basis of inherited disease, 2 ed., N. Y.- [a. o.], 1966. К. Д. Краснопольская. МОЛЕКУЛЯРНЫЕ И АТОМНЫЕ ПУЧКИ, направленные потоки молекул или атомов, движущихся в вакууме практически без столкновений друг с другом и с молекулами остаточных газов. М. и а. п. позволяют изучать свойства отд. частиц, пренебрегая эффектами, обусловленными столкновениями, кроме тех случаев, когда сами столкновения являются объектом исследований. Первый эксперимент с атомным пучком был осуществлён в 1911 франц. учёным Л. Дюнуайе, к-рый продемонстрировал прямолинейный пролёт в вакууме атомов Na. В дальнейшем эти эксперименты были продолжены О. Штерном с сотрудниками в Гамбурге (1929), к-рые использовали М. и а. п. для измерения скорости молекул и эффективных сечений их соударений друг с другом, а также для исследования явлений, обусловленных электронными спинами и магнитными моментами атомных ядер (см. Ядро атомное). В 1937 И. Раби использовал М. и а. п. в изобретённом им резонансном методе, к-рый вначале применялся для измерения магнитных моментов ядер (1937-40), а в дальнейшем стал осн. методом радиоспектроскопии, позволившим измерить с большой точностью фундаментальные характеристики молекул, атомов и атомных ядер (Н. Рамзей и др.). Рис. 1. Схема опыта для изучения химических реакций, происходящих при пересечении пучка атомов водорода с пучком двухатомных молекул щелочного металла. К1, К2, К3 - коллимнрующие щели. Источник, в к-ром формируются М. и а. п., представляет собой камеру, соединённую с высоковакуумным объёмом при помощи отверстия в тонкой стенке или узкого капилляра в толстой стенке. Исследуемые молекулы или атомы вводятся в камеру источника в виде газа или пара при давлении неск. мм рт. ст. Для формирования М. и а. п. давление газа в источнике должно быть достаточно малым, чтобы ср. длина I свободного пробега частиц внутри источника была равна или несколько больше диаметра соединит, отверстия. В этом случае частицы вылетают из источника независимо друг от друга. Для капилляра длина l должна быть соизмерима также с длиной капилляра. Чрезмерное увеличение l за счёт уменьшения давления в источнике, не улучшая существенно свойств М. и а. п., уменьшает их интенсивность. Для увеличения интенсивности пучков применяют источники с неск. отверстиями или капиллярами, расстояние между к-рыми должно быть несколько больше их диаметра. Соударения с частицами остаточного газа разрушают М. и а. п. тем быстрее, чем хуже вакуум. Длина М. и а. п. в идеальном вакууме была бы чрезвычайно велика, т. к. возможны были бы только соударения "догона". Молекулярное взаимодействие. Метод М. и а. п. даёт возможность детально изучать акт столкновения между двумя частицами, в отличие от химич. и газоди-намич. методов, в к-рых из-за множественных столкновений частиц друг с другом наблюдаются лишь усреднённые эффекты . В нек-рых из этих экспериментов измеряются эффективные сечения упругих и неупругих соударений частиц, движущихся под разными углами и с разными скоростями. В др. экспериментах наблюдаются химич. реакции между частицами и изучается угловое и энергетич. распределение продуктов реакции (Лестер, 1971; Дж.Росс, 1966; Р. Дж. Горд он и др., 1971). Типичный эксперимент второго рода показан на рис. 1. Атомы водорода вылетают из источника в вакуумную камеру, где они сталкиваются с двухатомными молекулами щелочного металла, напр. К2- Угловое распределение продуктов реакции измеряется с помощью детекторов с поверхностной ионизацией (горячие нити Pt и W). Т. к. вольфрамовый детектор одинаково чувствителен к частицам К3 и КОН, а платиновый - менее чувствителен к КОН, то, комбинируя оба детектора, можно различать эти молекулы. Иногда М. и а. п. предварительно поляризуют или, наоборот, измеряют появляющуюся поляризацию. В нек-рых экспериментах исследуется возбуждение колебательных уровней энергии у продуктов реакции. Резонансные эксперименты (метод Раби). Частицы, вылетая из источника в
вакуум (13,3 мн/м2 или 10-7 мм рт. ст.), пролетают
через неоднородное магнитное поле, создаваемое магнитом А (рис. 2).
Неоднородное поле А искривляет их траектории, что обусловлено
взаимодействием их магнитных моментов с неоднородным магнитным полем. Далее
частицы пролетают через коллиматор и попадают в область детектора, где
происходит компенсация искривления траектории в неоднородном магнитном поле,
создаваемом магнитом В. Конфигурация поля В в точности
противоположна конфигурации поля А. Для индентификации молекул их
ионизируют (электронным ударом) и пропускают через масс-спектрометр, после
чего они регистрируются электронным умножителем, соединённым с
фазочувствительным детектором. Плавно изменяя частоту v колебаний
электромагнитного поля в зазоре магнита С, создающего однородное магнитное поле,
измеряют интенсивность пучка, регистрируемого детектором. Если частота v
удовлетворяет воровскому условию: где п - Планка постоянная, то молекулы под действием электромагнитного поля, возбуждаемого в резонаторе Р, могут переходить из состояния с энергией "Л в состояние с энергией Е2 и обратно. Рис. 2. Схема эксперимента по наблюдению магнитного резонанса в молекулярном пучке. Пролёт частицы через прибор определяется по искривлению её траектории; отклонения увеличены относительно типичных размеров прибора (длина прибора 3 м, максимальное поперечное сечение 0,01 см). Р - резонатор, в котором возбуждается электромагнитное поле резонансной частоты; H1 - форвакуумный насос. Н2 - высоковакуумный насос; А, В и С - электромагниты. Если по магнитным свойствам состояние Е1отличается от состояния Е2, то поле В после перехода молекулы обычно компенсирует отклонение, вызванное полем А, не для всех молекул пучка; часть молекул, испытавшая переход Е1i->Е2> движется по траектории, показанной пунктиром (рис. 2). При выполнении условия (1) интенсивность, регистрируемая детектором, имеет минимум. График зависимости интенсивности от частоты представляет собой радиочастотный спектр частиц. Зная резонансную частоту из условия (1), можно определить уровни энергии молекул (см. Магнитный резонанс). Метод пароэлектрического резонанса аналогичен методу магнитного резонанса за исключением того, что изменения траектории обусловлены взаимодействием электрич. моментов молекул с неоднородными электрич. полями, а квантовые переходы между ними вызваны колебаниями электрич. поля в резонаторе. Интенсивность пучка может быть увеличена за счёт использования 4-полюсных или 6-полюс-ных электродов, создающих пространственную фокусировку пучка. Применяется также сочетание обоих методов, напр, однородное постоянное электрич. поле используют в экспериментах с магнитным резонансом, а однородное магнитное поле в опытах с параэлектрич. резонансом (К. Мак-Адан, Н. Рамзей и др., 1972). Эксперименты с магнитным и параэлект-рическим резонансами в М. и а. п. дали большое количество информации о строении молекул, атомов и атомных ядер. Этим методом были измерены спины ядер, магнитные и электрические квадрупольные моменты стабильных и радиоактивных ядер. В частности, был обнаружен электрич. квадрупольный момент дейтрона, что впервые указало на существование тензорных сил между элементарными частицами. Была измерена с высокой точностью тонкая структура атомных спектров, в результате чего в экспериментах с атомарным водородом был открыт Лэмбовский сдвиг, послуживший источником серии революц. теоретич. открытий в квантовой электродинамике. Измерения сверхтонкой структуры спектров дали первые указания на аномальность магнитного момента электрона, к-рая впоследствии была измерена непосредственно. В экспериментах с М. и а. п. были осуществлены два независимых измерения постоянной тонкой структуры и получено пока единственное доказательство существования у ядер электрич. октупольных моментов. Резонансные эксперименты с М. и а. п. позволили измерить вращательные магнитные моменты и электрич. дипольные моменты молекул, энергию взаимодействия ядерных магнитных моментов с вращательными магнитными моментами молекул, зависимость электрических и магнитных свойств от ориентации молекул; определить квадрупольные моменты молекул, энергию межъядерных магнитных взаимодействий в молекулах и т. д. Частота колебаний, соответствующая линиям сверхтонкой структуры магнитного резонанса в М. и а. п., является основой для определения секунды в пассивных стандартах частоты (см. Квантовые стандарты частоты, Квантовые часы). Возможность пространственной фокусировки М. и а. п., содержащих частицы в определённых энергетич. состояниях при помощи неоднородных электрических или магнитных полей, позволила использовать М. и а. п. для накопления частиц в состояниях с более высокой энергией (т. е. для создания инверсии населённостей), что необходимо для осуществления мазера. Первый мазер был осуществлён на пучке молекул аммиака (см. Молекулярный генератор). Мазер на пучке атомов водорода широко использовался как для исследования атома водорода, так и для создания активного квантового стандарта частоты. Лит.: Смит К. Ф-, Молекулярные пучки, пер. с англ., М., 1959; Рамзей Н., Молекулярные пучки, пер. с англ., М., I960; Kusch P., Huges V. W., Atomic and molecular beam spectroscopy, в кн.: Handbuch der Physik, Hrsg. von S. Flügge, Bd 37, Tl 1, В.- [u.a.l, 1959; Zorn J. C., English Т. С., Methods of experimental physics, v. 3, N. Y., 1973. Я. Ф. Рамзей (США). МОЛЕКУЛЯРНЫЕ КРИСТАЛЛЫ, кристаллы, образованные из молекул, связанных друг с другом слабыми ван-дер-ваальсовыми силами (см. Межмолекулярное взаимодействие) или водородной связью. Внутри молекул между атомами действует более прочная ковалентная связь. Фазовые превращения М. к.- плавление, возгонка, полиморфные переходы (см. Полиморфизм) - происходят, как правило, без разрушения отд. молекул. Большинство М. к.- кристаллы орга-нич. соединений, типичный М. к.- нафталин. М. к. образуют также нек-рые простые вещества (Н2, галогены, N2, O2, S8), бинарные соединения типа H2jO, СО2, N2O4, металлоорганические соединения и нек-рые комплексные соединения. КМ. к. относятся и кристаллы полимеров, а также кристаллы белков, нуклеиновых кислот. Особым случаем М. к. являются кристаллы отвердевших инертных газов, в которых ван-дер-ваальсовы силы связывают между собой не молекулы, а атомы. Для типичных М. к. характерны низкие температуры плавления, большие коэфф. теплового расширения, высокая сжимаемость, малая твёрдость. В обычных условиях большинство М. к. - диэлектрики. Нек-рые М. к., напр, органические красители, -полупроводники. Лит.: Китайгородский А. И., Молекулярные кристаллы, М., 1971; Бокий Г. Б., Кристаллохимия, М., 1971. П. М. Зоркий. МОЛЕКУЛЯРНЫЕ СИТА, сорбенты, избирательно поглощающие из окружающей среды вещества, молекулы к-рых не превышают определённых размеров. Такие сорбенты как бы отсеивают крупные молекулы от мелких. Различают минеральные (неорганические) и органич. М. с. Неорганич. М. с. имеют жёсткую кристаллич. структуру, в к-рой находятся полости, соединённые между собой узкими каналами-опорами" или "окнами". Малые размеры "окон" препятствуют диффузии крупных молекул во внутр. полости сорбента. Нек-рые алюмосиликаты - природные и синтетич. цеолиты - характерные представители М. с. этого типа. Органич. М. с.- гелевидные сорбенты, получаемые на основе высокомолекулярных соединений. Структура таких сорбентов представляет собой пространственную сетку из цепочечных макромолекул, "сшитых" в отд. точках химич. связями. Из гелевидных М. с. пром. производства наиболее распространены различные типы сефадекса - сорбента на основе декстрана (высокомолекулярного полисахарида). М. с., содержащие ионогенные (диссоциирующие на ионы) группы и способные к ионному обмену, наз. ионитовыми ситами. В отличие от обычных ионитов, они избирательно поглощают из раствора лишь достаточно малые ионы, исключая из ионообменного процесса крупные ионы, диффузия к-рых сквозь структурную сетку сорбента затруднена. М. с. выпускают в виде порошка, зёрен неправильной формы, сферич. гранул. Их используют для очистки веществ от нежелательных примесей, фракционирования синтетич. полимеров, хрома-тографич. разделения белков, углеводов, гормонов, антибиотиков и пр. Лит.: Детерман Г., Гель-хроматография, пер. с нем., М., 1970. Л. А. Шиц. МОЛЕКУЛЯРНЫЕ СПЕКТРЫ, оптические спектры испускания и поглощения, а
также комбинационного рассеяния света, принадлежащие свободным или слабо
связанным между собой молекулам. М. с. имеют сложную структуру. Типичные
М. с.- полосатые, они наблюдаются в испускании и поглощении и в комбинационном
рассеянии в виде совокупности более или менее узких полос в ультрафиолетовой,
видимой и близкой инфракрасной областях, распадающихся при достаточной
разрешающей силе применяемых спектральных приборов на совокупность тесно
расположенных линий. Конкретная структура М. с. различна для различных молекул
и, вообще говоря, усложняется с увеличением числа атомов в молекуле. Для весьма
сложных молекул видимые и ультрафиолетовые спектры состоят из немногих широких
сплошных полос; спектры таких молекул сходны между собой. М. с. возникают при квантовые
пере- где hv - энергия испускаемого или поглощаемого фотона частоты v (h -Планка постоянная). При комбинационном рассеянии hv равно разности энергий падающего и рассеянного фотонов. М. с. гораздо сложнее линейчатых атомных спектров, что определяется большей сложностью внутр. движений в молекуле, чем в атомах. Наряду с движением электронов относительно двух или более ядер в молекулах происходят колебательное движение ядер (вместе с окружающими их внутр. электронами) около положений равновесия и вращательное движение молекулы как целого. Этим трём видам движений - электронному, колебательному и вращательному - соответствуют три типа уровней энергии и три типа спектров. Согласно квантовой механике, энергия всех видов движения в молекуле может
принимать лишь определённые значения, т. е. она квантуется. Полная энергия
молекулы $ приближённо может быть представлена в виде суммы квантованных
значений энергий трёх видов её движения: му электронному состоянию соответствуют определённая равновесная конфигурация и определённое значение Еэл+, наименьшее значение соответствует осн. уровню энергии. Набор электронных состояний молекулы определяется свойствами её электронной оболочки. В принципе значения Еэл можно рассчитать методами квантовой химии, однако данная задача может быть решена только с помощью приближённых методов и для сравнительно простых молекул. Важнейшую информацию об электронных уровнях молекулы (расположение электронных уровней энергии и их характеристики), определяемую её хим. строением, получают, изучая её М. с. Рис. 1. Схема уровней энергии двухатомной молекулы: а и б -электронные уровни; v' и v" - квантовые числа колебательных уровней; J' и J" -квантовые числа вращательных уровней. Весьма важная характеристика заданного электронного уровня энергии -значение квантового числа S, характеризующего абс. величину полного спинового момента всех электронов молекулы. Химически устойчивые молекулы имеют, как правило, чётное число электронов, и для них S = 0,1,2... (для осн. электронного уровня типично значение S = 0, а для возбуждённых - S = 0 и S = 1). Уровни с S = 0 наз. синглетными, с S = 1 - триплетными (т. к. взаимодействие в молекуле приводит к их расщеплению на х = = 2S + 1 = 3 подуровня; см. Мультиплетность). Радикалы свободные имеют, как правило, нечётное число электронов, для них S = ½, 3/2, ... и типично как для основного, так и для возбуждённых уровней значение S = ½ (дублетные уровни, расщепляющиеся на и = 2 подуровня). Для молекул, равновесная конфигурация к-рых обладает симметрией, электронные уровни можно дополнительно классифицировать. В случае двухатомных и линейных трёхатомных молекул, имеющих ось симметрии (бесконечного порядка), проходящую через ядра всех атомов (см. рис. 2, б), электронные уровни характеризуются значениями квантового числа X, определяющего абс. величину проекции полного орбитального момента всех электронов на ось молекулы. Уровни с лямбда = 0,1,2,... обозначаются соответственно сумма, П, Д, ..., а величина и указывается индексом слева вверху (напр., 3сумма, 2п, ...). Для молекул, обладающих центром симметрии, напр. СО2 и С6Н6 (см. рис. 2, б, в), все электронные уровни делятся на чётные и нечётные, обозначаемые индексами g и и (в зависимости от того, сохраняет ли волновая функция знак при обращении в центре симметрии или меняет его). Колебательные уровни энергии (значения Екол) можно найти квантованием
колебательного движения, к-рое приближённо считают гармоническим. В простейшем
случае двухатомной молекулы (одна колебательная степень свободы,
соответствующая изменению межъядерного расстояния r) её рассматривают как
гармонич. осциллятор; его квантование даёт равноотстоящие уровни
энергии: где ve - осн. частота гармонич. колебаний молекулы, v - колебательное квантовое число, принимающее значения О, 1, 2, .... На рис. 1 показаны колебательные уровни для двух электронных состояний. Для каждого электронного состояния многоатомной молекулы, состоящей из N атомов
(N => 3) и имеющей f колебательных степеней свободы (f =
= 3N - 5 и f = 3N - 6 для линейных и нелинейных молекул
соответственно), получается f т. н. нормальных колебаний с частотами vi
(i = = l,2,3,...,f ) и сложная система колебательных уровней: где vi = О, 1, 2, ... - соответствующие колебательные квантовые числа. Набор частот нормальных колебаний в основном электронном состоянии является очень важной характеристикой молекулы, зависящей от её хим. строения. В определённом нормальном колебании участвуют все атомы молекулы или часть их; атомы при этом совершают гармонич. колебания с одной частотой vi, но с различными амплитудами, определяющими форму колебания. Нормальные колебания разделяют по их форме на валентные (при к-рых изменяются длины линий связи) и деформационные (при к-рых изменяются углы между хим. связями - валентные углы). Число различных частот колебаний для молекул низкой симметрии (не имеющих осей симметрии порядка выше 2) равно 2, и все колебания являются невырожденными, а для более симметричных молекул имеются дважды и трижды вырожденные колебания (пары и тройки совпадающих по частоте колебаний). Напр., у нелинейной трёхатомной молекулы Н2О (рис. 2, a) f = 3 и возможны три невырожденных колебания (два валентных и однс деформационное). Более симметричная линейная трёхатомная молекула СО2 (рис. 2, б) имеет f = 4 - два невырожденных колебания (валентных) и однс дважды вырожденное (деформационное). Для плоской высокосимметричной молекулы С6Н6 (рис. 2, в) получается f = 30 - десять невырожденных и 1C дважды вырожденных колебаний; из них 14 колебаний происходят в плоскости молекулы (8 валентных и 6 деформационных) и 6 неплоских деформационных колебаний - перпендикулярно этой плоскости. Ещё более симметричная тетраэдрическая молекула СН4 (рис. 2, г имеет f = 9 - одно невырожденное колебание (валентное), одно дважды вырожденное (деформационное) и два трижды вырожденных (одно валентное и одно деформационное). Рис. 2. Равновесные конфигурации молекул: а - H2О; б - СО2; в - С6Н6; г - СН4. Числами указаны длины связей (в А) и величины валентных углов. Вращательные уровне энергии можно найти квантованием вращательного движения
молекулы, рассматривая её как твёрдое тело с определенными моментами
инерции. В простейшем случае двухатомной или линейной многоатомной
молекулы её энергии вращения где I - момент инерции молекулы относительно оси, перпендикулярной оси
молекулы, а М - вращательный момент количества движения. Согласно
правила квантования. определяет масштаб расстоянии между уровнями энергии, уменьшающийся с увеличением масс ядер и межъядерных расстояний. На рис. 1 показаны вращательные уровни для каждого электронно-колебательного состояния. Различные типы М. с. возникают npи различных типах переходов между уровнями энергии молекул. Согласно (1 и (2) Рис. 3. Электронно-колебательный спектр молекулы N2 в близкой ультрафиолетовой области; группы полос соответствуют различным значениям Д v= v' - v". Рис. 4. Вращательное расщепление электронно-колебательной полосы 3805 А молекулы N2. При ДЕэл = 0, а ДЕкол не= 0 получаются колебательные М. с., наблюдаемые в близкой (до неск. мкм) и в средней (до неск. десятков мкм) инфракрасной (ИК) области, обычно в поглощении, а также в комбинационном рассеянии света. Как правило, одновременно Д^вращ^ 0 и при заданном $кол получается колебательная полоса, распадающаяся на отдельные вращательные линии. Наиболее интенсивны в колебательных М. с. полосы, соответствующие Ди = v' - v" = 1 (для многоатомных молекул - Дvi" = vi" - vi" = 1 при Дvk= = vt,' - vk" = 0, где к не= i)/ Для чисто гармонич. колебаний эти отбора правила, запрещающие др. переходы, выполняются строго; для ангармонич. колебаний появляются полосы, для к-рых Дv > 1 (обертоны); их интенсивность обычно мала и убывает с увеличением Av. Колебательные (точнее, колебательно-вращательные) спектры изучаются экспериментально в ИК-ооласти в поглощении при помощи ИК-спектрометров с призмами, прозрачными для ИК-излучения, или с дифракционными решётками, а также Фурье-спектрометров и в комбинационном рассеянии при помощи светосильных спектрографов (для видимой области) с применением лазерного возбуждения. При ДЕэл = 0 и ДЕкол = 0 получаются чисто вращательные М. с., состоящие из отд. линий. Они наблюдаются в поглощении в далёкой (сотни мкм) ИК-области и особенно в микроволновой области, а также в спектрах комбинационного рассеяния. Для двухатомных и линейных многоатомных молекул (а также для достаточно симметричных нелинейных многоатомных молекул) эти линии равно отстоят (в шкале частот) друг от друга с интервалами Дv = 2В в спектрах поглощения и Дv = 4В в спектрах комбинационного рассеяния. Чисто вращательные спектры изучают в поглощении в далёкой ИК-области при помощи ИК-спектрометров со спец. дифракционными решётками (эшелеттами) и Фурье-спектрометров, в микроволновой области при помощи микроволновых (СВЧ) спектрометров (см. Микроволновая спектроскопия), а также в комбинационном рассеянии при помощи светосильных спектрографов. Методы молекулярной спектроскопии, основанные на изучении М. с., позволяют решать разнообразные задачи химии, биологии и др. наук (напр., определять состав нефтепродуктов, полимерных веществ и т. п.). В химии по М. с. изучают структуру молекул. Электронные М. с. дают возможность получать информацию об электронных оболочках молекул, определять возбуждённые уровни и их характеристики, находить энергии диссоциации молекул (по схождению колебательных уровней молекулы к границам диссоциации). Исследование колебательных М. с. позволяет находить характеристические частоты колебаний, соответствующие определённым типам хим. связей в молекуле (напр., простых двойных и тройных связей С - С, связей С - Н, N - Н, О - Н для органических молекул), различных групп атомов (напр., СН2, СН3, NH2), определять пространственную структуру молекул, различать цис- и трансизомеры. Для этого применяют как инфракрасные спектры поглощения (ИКС), так и спектры комбинационного рассеяния (СКР). Особенно широкое распространение получил метод ИКС как один из самых эффективных оптич. методов изучения строения молекул. Наиболее полную информацию он даёт в сочетании с методом СКР. Исследование вращательных М. с., а также вращательной структуры электронных и колебательных спектров позволяет по найденным из опыта значениям моментов инерции молекул [к-рые получаются из значений вращательных постоянных, см. (7)] находить с большой точностью (для более простых молекул, например Н2О) параметры равновесной конфигурации молекулы - длины связей и валентные углы. Для увеличения числа определяемых параметров исследуют спектры изотопических молекул (в частности, в которых водород заменён дейтерием), имеющих одинаковые параметры равновесных конфигураций, но различные моменты инерции. В качестве примера применения М. с. для определения хим. строения молекул рассмотрим молекулу бензола С6Н6. Изучение её М. с. подтверждает правильность модели, согласно к-рой молекула плоская, а все 6 связей С - С в бензольном кольце равноценные и образуют правильный шестиугольник (рис. 2, в), имеющий ось симметрии шестого порядка, проходящую через центр симметрии молекулы перпендикулярно сё плоскости. Электронный М. с. поглощения С6Н6 состоит из неск. систем полос, соответствующих переходам из основного чётного синглетного уровня на возбуждённые нечётные уровни, из к-рых первый является триплетным, а более высокие - синглетными (рис. 5). Наиболее интенсивна система полос в области 1840А (Е5 -Е5= 7,0 эв), наиболее слаба система полос в области 3400А (Е2 - Е1 = = 3,8 эв), соответствующая синглетно-триплетному переходу, запрещённому приближёнными правилами отбора для полного спина. Переходы соответствуют возбуждению т. н. я-электронов, делокализованных по всему бензольному кольцу (см. Молекула); полученная из электронных молекулярных спектров схема уровней рис. 5 находится в согласии с приближёнными квантовомеханическими расчётами. Колебательные М. с. С6Н6 соответствуют наличию в молекуле центра симметрии - частоты колебаний, проявляющиеся (активные) в ИКС, отсутствуют (неактивные) в СКР и наоборот (т. н. альтернативный запрет). Из 20 нормальных колебаний СбН6 4 активны в ИКС и 7 активны в СКР, остальные 11 неактивны как в ИКС, так и в СКР. Значения измеренных частот (в см~1): 673, 1038, 1486, 3080 (в ИКС) и 607, 850, 992, 1178, 1596, 3047, 3062 (в СКР). Частоты 673 и 850 соответствуют неплоским колебаниям, все остальные частоты - плоским колебаниям. Особо характерны для плоских колебаний частота 992 (соответствующая валентному колебанию связей С - С, состоящему в периодич. сжатии и растяжении бензольного кольца), частоты 3062 и 3080 (соответствующие валентным колебаниям связей С - Н) и частота 607 (соответствующая деформационному колебанию бензольного кольца). Наблюдаемые колебательные спектры С6Н6 (и аналогичные им колебательные спектры C6D6) находятся в очень хорошем согласии с теоретич. расчётами, позволившими дать полную интерпретацию этих спектров и найти формы всех нормальных колебаний. Рис. 5. Схема электронных уровней и переходов для молекулы бензола. Энергия уровней дана в эв. С - синглетные уровни; Т - триплетный уровень. Чётность уровня указана буквами g и и. Для систем полос поглощения указаны примерные области длин волн в А, более интенсивные системы полос обозначены более жирными стрелками. Подобным же образом можно при помощи М. с. определять структуру разнообразных классов органических и неорганических молекул, вплоть до весьма сложных, напр, молекул полимеров. Лит.: Кондратьев В. Н., Структура атомов и молекул, 2 изд., М., 1959; Ельяшевич М. А., Атомная и молекулярная спектроскопия, М., 1962; Г е р ц б е р г Г., Спектры и строение двухатомных молекул, пер. с англ., М., 1949; его же, Колебательные и вращательные спектры многоатомных молекул, пер. с англ., М., 1949; его же, Электронные спектры и строение многоатомных молекул, пер. с англ., М., 1969; Применение спектроскопии в химии, под ред. В. Веста, пер. с англ., М., 1959. М. А. Ельяшевич. |
|
© (составление) libelli.ru 2003-2020 |